13.03.2019
Boli limfoproliferative.
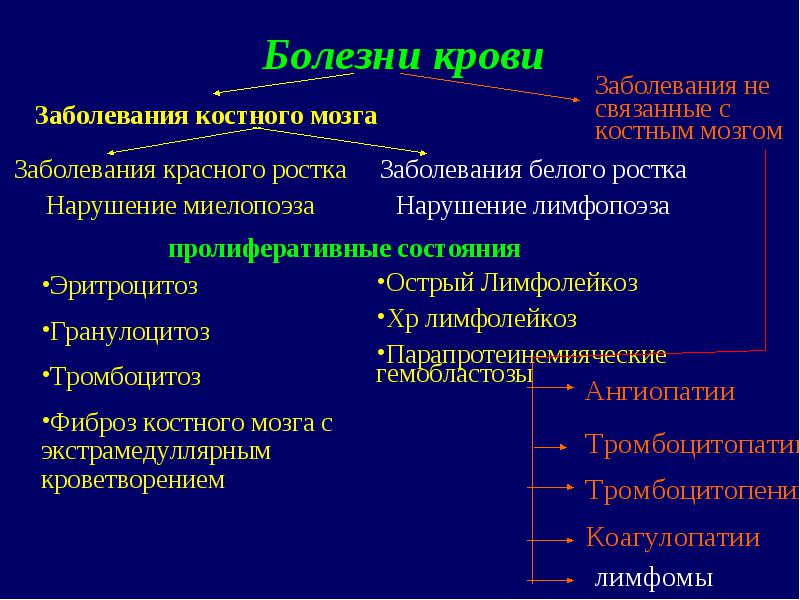
Boli ale sângelui Boli ale germenului roșu Tulburări mielopoiezei Boli ale germenilor albi Tulburări limfopoiezei Eritrocitoză Granulocitoză Trombocitoză Fibroză măduvă osoasă cu afectiuni proliferative de hematopoieza extramedulara Boli ale maduvei osoase Boli care nu sunt legate de maduva osoasa Leucemie limfocitara acuta Leucemie limfocitara chr Hemoblastoze paraproteinemice Angiopatie limfom Trombocitopatie Trombocitopenie Coagulopatie
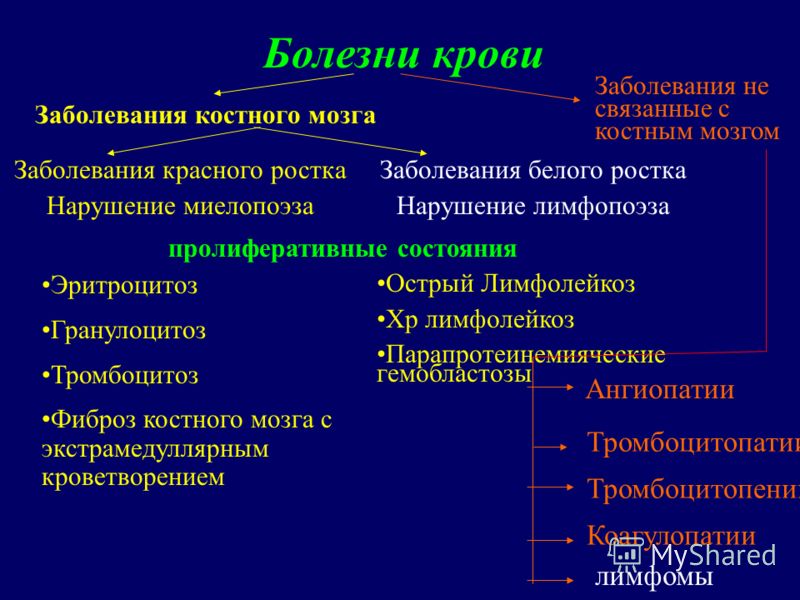
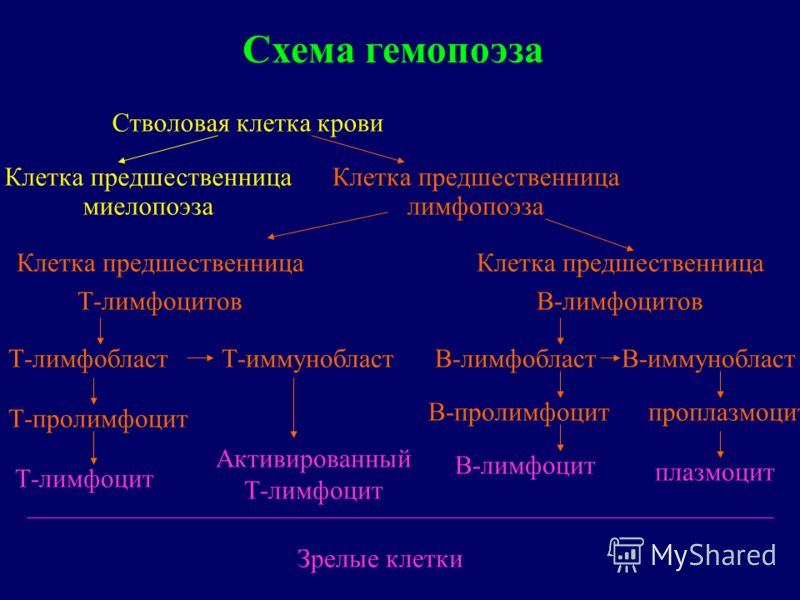
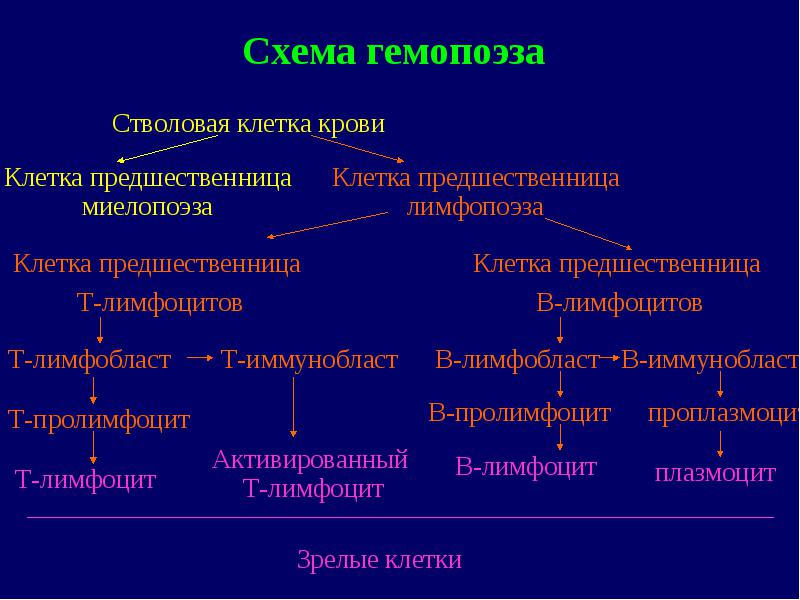
Diagrama hematopoiezei Limfociteza celule progenitoare limfocite T celula progenitoare limfocite B celule progenitoare limfocite T prolimfocite T limfocite T limfocite T activate limfocite B limfoblast B prolimfocite B prolimfocite B-prolimfocite plasmatice B-limfoblaste plasme celulă stem Celulele sanguine precursoare ale mielopoiezei
Clasificarea euro-americană a bolilor limfoproliferative (după N.L. Harris et al., 1994). Tumorile cu celule B. I. Tumori de la progenitorii precoce ai limfocitelor B: -- leucemie/limfom de la progenitorii limfoblastelor B. II. Tumori cu celule B periferice. 1. Leucemie limfocitară cronică cu celule B / leucemie prolimfocitară / limfom limfocitar mic. 2. Limfom/imunocitom limfoplasmocitar. 3. Limfom din celulele mantalei. 4. Limfom din centrul folicular, categorii citologice preliminare foliculare: I-- celule mici; II - celule mixte mici și mari; III - celula mare; 5. Limfomul cu celule B al zonei marginale. 6. Limfom al splinei, originar din zona marginală 7. Leucemie cu celule piloase. 8. Plasmacitom / mielom cu celule plasmatice. 9. Limfom difuz din celulele B mari. 10. Limfomul Burkitt. 11. Tip provizoriu: limfom cu celule B grad înalt malignitate, asemănătoare Burkitt.

Clasificarea euro-americană a bolilor limfoproliferative (după N.L. Harris et al., 1994). Tumori cu celule T și tumori natural killer. I. Tumora de la progenitorii timpurii ai celulelor T: -- leucemie/limfom de la progenitorii limfoblastelor T II. Tumori periferice cu celule T și tumori natural killer 1. Leucemie limfocitară cronică cu celule T / leucemie prolimfocitară. 2. Leucemie din limfocitele granulare mari. -- tipul celulelor T; -- tipul celulei NK; 3. Micoza fungică / sindromul Cesari. 4. Limfom periferic cu celule T, 5. Limfom angioimunoblastic cu celule T. 6. Limfom angiocentric. 7. Limfom cu celule T intestinal (+/- asociat cu enteropatie). 8. Leucemie/limfom cu celule T la adulți. 9. Limfom anaplazic cu celule mari, tipuri CD30+, T- și celule nule. 10. Tip provizoriu: limfom anaplazic cu celule mari, asemănător Hodgkin. boala Hodgkin (limfogranulomatoza). I. Predominanța limfoidă. II. scleroza nodulara. III. Varianta mixta. VI. Depleția limfoidă.

Grupul bolilor limfoproliferative include: 1. Leucemia limfoblastică acută 2. Leucemie limfocitară cronică 3. Hemoblastoze paraproteinemice 4. Limfogranulomatoza (limfom Hodgkin) 5. Limfoame non-Hodgkin (limfosarcoame) LPZ - neoplasme maligne sau benigne care se dezvolta din celulele limfoide situate pe diferite etape diferenţiere.neoplasme maligne sau benigne ale celulelor limfoide

Limfoame non-Hodgkin (limfosarcoame) 1. Un grup eterogen de boli neoplazice care provin din sistem imunitar 2. Sursa celulară a tumorii este o celulă a ganglionului limfatic periferic 3. Caracterizată printr-o creștere a ganglionilor limfatici și/sau afectarea diferitelor organe interne, în care are loc o acumulare necontrolată de limfocite „tumorale”.

Clasificarea Organizației Mondiale a Sănătății a limfoamelor non-Hodgkin Tumori cu celule B progenitoare: Limfom/leucemie limfoblastică B progenitoare (leucemie limfoblastică acută cu celule B progenitoare). Tumori cu celule B din limfocitele B periferice (mature): leucemie limfocitară cronică cu celule B/limfom limfocitar mic (limfom limfocitar) Leucemie prolimfocitară cu celule B Limfom limfoplasmocitar Limfom splenic din zonă marginală (+/- leucemie viloasă) limfom celulozic celular mielom/plasmocitom Limfom extranodal cu celule B din zona marginală de tip MALT Limfom ganglionar cu celule B din zona marginală (+/- limfocite B monocitoide) Limfom folicular Limfom cu celule de manta Limfom cu celule B difuze Limfom difuz cu celule B mari mediastinal Primar limfom exudativ Limfom / leucemie Burkitt

Clasificarea limfoamelor non-Hodgkin ale Organizației Mondiale a Sănătății tumorilor cu celule T și NK din limfocitele T progenitoare: limfom T limfoblastic/leucemie progenitoare (leucemie limfoblastică acută cu celule T progenitoare) Limfoame T cu celule T periferice (mature) limfocite: leucemie prolimfocitară cu celule T Leucemie cu celule T cu limfocite mari granulare Leucemie agresivă cu celule NK Limfom/leucemie cu celule T pentru adulți (HTLV1+) Limfom cu celule T extranodal NK/T, tip nazal Limfom cu celule T asociat enteropatiei Limfom cu celule T hepatolienal limfom limfom asemănător paniculitei cu celule T țesut subcutanat Micoză fungoide / sindrom Cesari Limfom anaplazic cu celule mari, cu celule T/0, cu leziune primară piele Limfom periferic cu celule T, nespecificat Limfom angioimunoblastic cu celule T Limfom anaplazic cu celule T/0 cu celule mari, cu afectare sistemică primară


Patogenia limfoamelor 1. Creșterea și metabolismul tumorii 2. Celulele tumorale inhibă dezvoltarea celulelor normale și provoacă deficiență imunologică ( stare de imunodeficiență). 3. Dezvoltați reacții imune cauzate de producerea de anticorpi direcționați împotriva antigenelor proprii țesuturilor (anemie hemolitică imună sau trombocitopenie imună, aplazie parțială a celulelor roșii la pacienții cu limfom) 4. Funcția organelor din apropiere este afectată

Tabloul clinic al limfoamelor Simptome de intoxicație - nu provoacă senzații subiective la pacient și poate fi detectat în timpul unei examinări ocazionale - slăbiciuni, oboseală, cresterea temperaturii, pierdere în greutate - transpirații abundente, în special noaptea, mâncărimi ale pielii nemotivate și toleranță slabă la mușcăturile de insecte care suge sânge Simptome de progresie tumorală - Sindrom metastatic (l/a mare) - Sindrom imunologic (anemie hemolitică imună, trombocitopenie imună, sindrom asemănător lupusului) - Scăderea stării imunitare (bacterie frecventă și infecții virale)



Diagnosticul limfoamelor Diagnosticul limfomului se bazează pe studiul substratului morfologic al tumorii - Biopsia (extirparea chirurgicală) a ganglionului afectat cu examenul morfologic și imunologic ulterioar.De obicei, punctul de plecare al unei căutări diagnostice este depistarea a unei mariri nemotivate a ganglionilor limfatici. Mărirea unui ganglion limfatic fără motiv aparent la o dimensiune mai mare de 1 cm și existența unui astfel de ganglion mărit mai mult de 1 lună reprezintă baza pentru efectuarea unei biopsii a ganglionului limfatic.


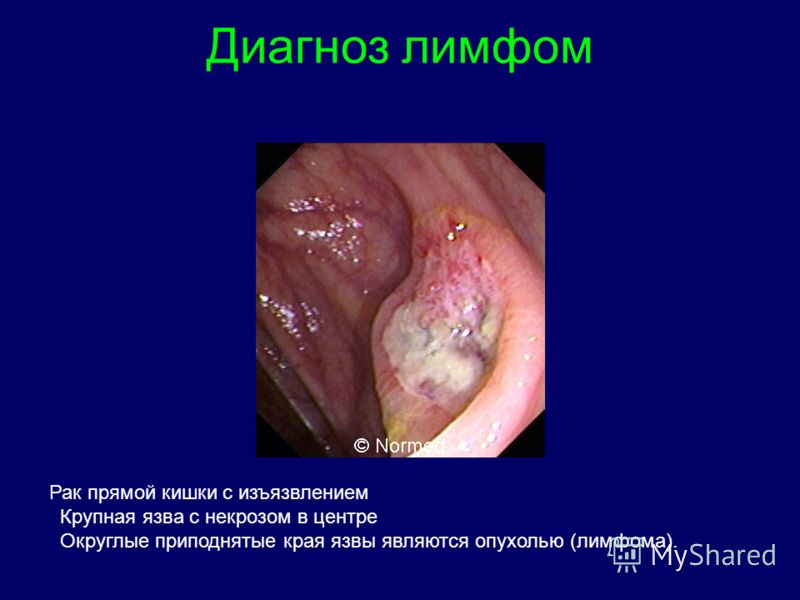
Clasificarea stadiului limfoamelor Stadiul I - o creștere a unui grup de ganglioni limfatici Etapa II - o creștere a două sau mai multe grupuri de ganglioni limfatici pe o parte a diafragmului Stadiul III - O creștere a două sau mai multe grupuri de ganglioni limfatici pe laturile opuse ale diafragmei Stadiul IV - Lezarea organelor interne Stadiul V - afectarea celulelor tumorale maduvei osoase "limfom cu leucemizare" Substadiul A B


O versiune simplificată a diagnosticului limfoamelor non-Hodgkin în funcție de gradul de malignitate: Limfoame formate din celule imature (limfoblaste) - limfom de grad înalt. Limfoame formate din celule de maturitate intermediară (prolimfocite) - limfom de grad intermediar de malignitate. Limfoamele formate din celule mature (limfocite) sunt limfoame de grad scăzut.


caracteristici generale boli limfoproliferative maligne. Leucemie limfobdastică acută Limfom Lymphogra nulomatosis Origine 80% celule B 20% celule T 90% celule B 10% celule T Nedeterminat celule T) Rare Frecvente Cavitate abdominală Rare Frecvente Rare Măduvă osoasă Întotdeauna frecvente Rar Prezența simptomelor generale Frecvente Rare Frecvente Aberații cromozomiale Frecvente (translocații, deleții) Frecvente (aneuploidii) Rata de vindecare 40-60% 30-40% 75-85%
Pacientul P., 72 de ani, a fost internat pentru exacerbare pneumonie cronică. La examinare, mărită la ou de gaina ganglioni limfatici: cervicali, inghinali, axilari. Ganglionii limfatici sunt moi, nu sunt lipiți între plămâni: o nuanță de sunet de percuție, împrăștiat uscat pe ambele părți. Hepatosplenomegalie. Sânge: Hb - 74 g/l, eritrocite - 2,3 1012, reticulocite - 20%, leucocite - 5,0 109: e - 1%, s/i - 2%, s/i - 17%, limfocite - 79%, monocite - 1%, VSH 60 mm/h, celule de leucoliză. Studiu de caz cu trombocite



Întrebări interactive 3. Dacă celulele morfologic asemănătoare cu celulele focarului primar al limfomului sunt detectate în punctatul măduvei osoase, ar trebui să ne gândim la 1. Transformarea limfomului în leucemie 2. Metastaza limfomului în măduva osoasă 3. Dezvoltarea unui blast criza limfomului


Întrebări interactive 5. Ce poate fi în măduva osoasă punctată cu limfomul Hodgkin 1. Compoziție celulară normală 2. Limfocitoză până la 20% 3. Celule similare morfologic cu celulele focarului primar al limfoamelor 4. Inhibarea tuturor germenilor hematopoietici 5. Exploziile sunt determinate


Literatură 1. Abdulkadyrov K.M. et al. Sindroame hematologice în general practica clinica// „Elbi”, Sankt Petersburg C Sindroame hematologice în practica clinică, editat de Vyagorskaya Ya.I., Kiev „Sănătate” 1981 od 3. Vorobyov A.I. Ghid de hematologie.// „Newdiamed”, Moscova T1. 4. Wood M.E., Bunn P.A. Secretele hematologiei și oncologiei // „Binom” - Moscova S. Guseva S.A., Voznyuk V.P. Boli ale sistemului sanguin. Director. // „MEDpress-inform”. - Moscova S Boli interne editat de Ryabva S.I., Almazov V.A., Shlyakhto E.V., Saint Petersburg, SpecLit, 2000 7. Clinical oncohematology, editat de Volkova M.A., Moscova, „Medicine”, 2001 8. Shiffman F.D. Fiziopatologia sângelui. // „Binom”.-Moscova S, M. Wetzler, K. Bloomfield LEUCEMIA MIELOIDĂ. Din Harrison’s Principles of Internal Medicine, ediția a XIV-a, G.I. Abelev, Mecanisme de diferențiere și creșterea tumorii. Biochimie, 2000, 65, Bolile sângelui, „editura enciclopediei”, Moscova, 2005 12. Diagnosticul și tratamentul sângelui DIC, Lychev V.G., Moscova, „medicina” 1993 13. Diagnosticul și terapia controlată a tulburărilor de hemostază Z, .S., Momot AP, Newdiamed, Moscova, 2001 14. Hemostaza reală, Vorobyov PA, Newdiamed, Moscova, 2004

Boli mieloproliferative și limfoproliferative: epidemiologie, clinică, tratament
UNIVERSITATEA MEDICALĂ DE STAT BELARUSIAN
ESEU
Pe subiect:
„BOLI MIELOPROLIFERATIVE ȘI LIMFOPROLIFERATIVE: EPIDIMIOLOGIE, CLINICĂ, TRATAMENT”
MINSK, 2008
BOLI MIELOPROLIFERATIVE (MIELOLEUCEMIE CRONICĂ, POLICITEMIE VERA)
Leucemia mielogenă cronică (LMC) este o tumoare care apare din celulele precursoare ale mielopoiezei timpurii care se diferențiază în forme mature.
Substratul celulei LMC:
predominant granulocite, în principal neutrofile.
EPIDEMIOLOGIE
LMC este un tip comun de leucemie (20%);
din punct de vedere al prevalenței, se situează pe locul 3 după leucemia acută și LLC;
incidență - 1-1,5 la 100 mii populație în ultimii 50 de ani;
bărbații reprezintă 55-60% dintre pacienți;
debutul bolii este la vârsta de 30-50 de ani;
la copii – rar (1-2%).
Printre influențele externe care contribuie la dezvoltarea LMC, rețineți:
IS (de exemplu, victime ale bombardamentelor de la Hiroshima și Nagasaki; pacienți cu spondiloartrită care au primit terapie cu raze X);
agenţi chimici (rolul benzenului dovedit).
PATOGENEZĂ
există un transfer al majorității brațului lung al cromozomului 22 la brațul lung al cromozomului 9 și o mică parte terminală a brațului lung al cromozomului 9 este transferată la cromozomul 22;
un cromozom de 22 de perechi cu un braț lung scurtat este desemnat ca cromozom Ph ("Philadelphia");
proto-oncogena ABL este situată pe brațul lung al cromozomului 9 (q34) (gena normală ABL codifică formarea unei proteine cu o greutate moleculară de 145 kDa, aparținând familiei tirozin protein kinazelor care sunt implicate în proces a fosforilării aminoacizilor în ciclul celular);
în timpul translocării (9;22), o parte a genei ABL este transferată de la cromozomul 9 la brațul lung al cromozomului 22 la locul unde a avut loc ruptura și este localizată gena BCR (produsul genei normale BCR este o proteină cu o greutate moleculară de 160 kDa; în absența genei BCR apar defecte funcționale neutrofile);
rezultatul acestei fuziuni este formarea pe cromozomul 22 a genei himerice BCR-ABL, care codifică o proteină himerică cu o greutate moleculară de 210 kDa, care are o activitate tirozin kinazei mai pronunțată decât prototipul său normal p145abl;
activarea diferitelor regiuni ale genei himerice determină un lanț de evenimente care conduc la o creștere a proliferării celulare.
CLINICA
Pe etape:
stadiu inițial: LMC practic nu este detectată (doar un studiu aleatoriu al CBC poate dezvălui modificări: leucocitoză, asociere bazofil-eozinofilă), nu există clinică;
stadiul de accelerare (manifestări detaliate): oboseală rapidă, transpirații, febră scăzută, scădere în greutate, greutate și durere în hipocondrul stâng cu splina mărită (poate fi chiar și în pelvisul mic), există infarcturi cu dureri ascuțite, ficatul poate fi ușor mărit, l / y practic nu sunt măriți, sindromul hemoragic este de obicei absent, în plămâni - pneumonie asociată cu infiltrație leucemică și infecție secundară, inimă: pot apărea aritmii; diagnosticare în această etapă:
modificări caracteristice ale KLA: anemie normocromă de severitate moderată (CP = 0,9), leucocitoză, o creștere a bazofilelor și eozinofilelor, o scădere a limfocitelor și puțin - monocite, o accelerare a VSH, trombocitele sunt reduse, dar fără sindrom hemoragic; neutrofile: sunt prezente toate formele (mieloblaste, promielocite, juvenili, bastonașe, segmente, fără dip leucemic);
CM punctat: o creștere a numărului de megacariocite, un procent crescut de granulocite imature cu o creștere a raportului mieloid-eritroid la 20-25: 1 (normal - 3-4: 1);
scăderea fosfatazei alcaline a neutrofilelor mai mică de 25 de unități;
detectarea cromozomului Ph în celulele hematopoietice din seria mieloide;
biochimie: o creștere a vitaminei B12 de 10-15 ori, o creștere a acidului uric (sindrom hiperuricemic), poate exista o creștere semnificativă a LDH.
stadiu terminal (criza blastica): caracterizat prin aparitia unor anomalii cromozomiale suplimentare in celulele maduvei osoase; în perioada de remisiune a crizei blastice, aceste anomalii cromozomiale suplimentare dispar; deteriorarea sănătății, febră persistentă, epuizarea pacientului, mărirea splinei și, într-o măsură mai mică, ficatul, modificări distrofice ale organelor interne, refractaritate la terapia în curs, poate apărea sindromul hemoragic, complicații infecțioase frecvente, boala capătă caracteristicile de leucemie acută.
Reacție leucemoidă - un răspuns excesiv la orice stimul, manifestat prin hiperleucocitoză și apariția unui exces de celule imature în sângele periferic și care dispar după eliminarea cauzei. Apare în următoarele condiții:
tuberculoză;
boli purulent-septice;
hepatită virală, ciroză hepatică;
tumori maligne;
boală medicinală;
arsuri severe;
intoxicație cu mercur.
Cu reacții leucemoide:
nu există asociere bazofil-eozinofilă;
activitate crescută a fosfatazei alcaline a neutrofilelor;
cromozomul Ph nu este niciodată detectat.
TRATAMENT LMC
TARGET-terapie (vedere);
Mielozanul (busulfan, mileran) din grupul medicamentelor alchilante a fost primul care a fost utilizat în tratamentul LMC;
in 1966, a aparut un mesaj despre hidroxiureea (hidroxiureea, hidrea, litalir), un inhibitor al ribonucleotidazei, o enzima necesara sintezei ADN-ului;
în anii 1980 o nouă eră în terapia LMC a început odată cu apariția α-interferonului; vă permite să obțineți remisie hematologică, citogenetică, scade Nivelurile LDHși vitamina B12;
citosar (Cytozar), Citozină-arabinozidă (Ara-C) este o nucleozidă pirimidină; metabolitul său activ inhibă ADN polimeraza, ceea ce duce la întreruperea sintezei ADN și la suprimarea creșterii celulelor Ph-pozitive;
în faza de accelerare și faza terminală se aplică schema 3 + 7 (rubomicină-antraciclină + citosar (ara-C)) la 1,5-2 luni;
medicamente noi în tratamentul LMC:
homoharringtonin - un analog sintetic al unui alcaloid din plante chinezești (+ ara-C în caz de rezistență la α-interferon);
decitabină - un inhibitor al hipermetilării în ciclurile celulare (acest proces se observă în timpul progresiei tumorilor, în perioada unei crize blastice);
acid all-trans-retinoic (ATRA - acid all-trans-retinoic) + IFNa;
topotecan - un inhibitor al enzimei topoizomerazei I, necesar pentru replicarea ADN-ului, + ara-C + ciclofosfamidă (în stadiul de accelerare și criză blastică);
GM-CSF (GM-CSF) + IFNa;
inhibitor mutant al tirozin kinazei: STI-571 (inhibitor de transducție a semnalului, inhibitor al căilor de transducție a semnalului), medicamentul Glivec (mosilat de imatinib), mecanismul său de acțiune: prin conectarea la centrii activi ai BCR-ABL-tirozin kinazei (proteina p210) , perturbă procesele de interacțiune a substraturilor din interiorul celulelor, ceea ce duce la moartea celulelor care conțin proteina p210, adică. Ph-pozitiv; tratamentul cu Gleevec depășește semnificativ toate abordările existente anterior în ceea ce privește posibilitatea restabilirii hematopoiezei Ph-negative;
MTC - în perioada de remisiune clinică și hematologică.
Policitemia vera (eritremia, boala Wakez) este o leucemie cronică cu leziune la nivelul celulei progenitoare mielopoiezei cu o proliferare nelimitată a acestei celule caracteristice unei tumori, păstrând în același timp capacitatea de diferențiere în trei muguri, în principal roșii.
În anumite stadii ale bolii, uneori încă de la început, metaplazia mieloidă din splină se alătură proliferării în măduva osoasă.
EPIDEMIOLOGIE
rata de incidență - 0,6-1,6 la 100 mii populație;
5-6 cazuri la 1 milion de locuitori pe an.
CLINICA
În tabloul clinic - 2 sindroame:
pletoric (pletoră - pletoră): o creștere a numărului de globule roșii, globule albe, trombocite în sânge;
mieloproliferativ: din cauza hiperplaziei tuturor celor trei linii hematopoietice din măduva osoasă și extramedulară.
Stadiile bolii:
Etapa I - inițială (5 ani sau mai mult):
pletora moderata, splina nu este palpabila, eritrocitoza moderata, panmieloza in CM, tensiunea arteriala poate fi crescuta, poate exista tromboza.
Stadiul IIA - eritremic (desfasurat) fara metaplazie mieloida a splinei:
o versiune simplă de pletoră fără mărirea splinei, durează 10-15 ani sau mai mult; afecțiunea este tulburată: dureri de cap de tipul migrenelor chinuitoare cu deficiență de vedere, dureri în inima tipului de angină pectorală, adesea mâncărimi ale pielii, eritromelalgie, (durere paroxistică arzătoare în vârful degetelor de la mâini și de la picioare cu înroșire a pielii ).
Etapa IIB - proces eritremic cu metaplazie mieloidă a splinei:
splenomegalie, hepatomegalie moderată, piele roșie-cianotică, tensiune arterială crescută destul de stabilă cu simptome cerebrale severe, tromboză a vaselor coronare și cerebrale, vaselor de sânge extremitati mai joase la clinica corespunzatoare pot aparea si sangerari (DIC cronica cu consumul) de la gingii, nas etc., apar (pentru prima data) ulcere gastrice si duodenale cu corespunzator. sindrom de durere, tulburări metabolice ale acidului uric (clinica de gută), urolitiază; în sânge: pancitoză, o abundență de celule sanguine înfundă patul microcirculator, începe epuizarea pacientului, șoc, reacții în organe și țesuturi cu pierderea funcției lor.
Stadiul III - degenerarea anemică terminală a unei tumori benigne într-una malignă:
apare o clinică tipică de LMC, care, la rândul său, dă o criză de explozie (caracteristici ale LMA); dacă rezultatul este mielofibroza (adică fibroza stromală a măduvei osoase) - o scădere a nivelului de eritrocite (anemie), trombocite, leucocite.
În KLA: o creștere a eritrocitelor, hemoglobinei, trombocitelor, leucocitelor, VSH este încetinită brusc (1 mm sau o liniuță), hematocritul 0,7.
Trepanobiopsia ilionului se efectuează în scopul confirmării morfologice a diagnosticului.
Eritrocitoza
| Forme de bază | Forme clinice |
| I. Absolut | |
| primar | eritremie |
| secundar: | |
| cauzate de hipoxie | „răul de altitudine”, boală pulmonară obstructivă, malformații congenitale „albastre”, sindromul Pickwick, carboxihemoglobinemie, hemoglobinopatii cu afinitate crescută pentru oxigen |
| asociat cu producția crescută de eritropoietină | cancer renal hipernefroid, hemangioblastom cerebelos, cancer hepatic, miom uterin, tumori ale corticalei și medulei suprarenale |
| II. relativ | pierderi de lichide de către organism (infecții, pancreatită etc.), tulburări emoționale, alcoolism, hipertensiune arterială sistemică |
Măsuri terapeutice pentru eritremie
| Direcții principale | Remedii |
| eliminarea multitudinii | sângerare (300-500 ml o dată la două zile până la Hb 150, după - reopoliglucină și agenți antiplachetari (trental) IV), agenți antiplachetari, eritrocitafereză (1-2 proceduri în 5-7 zile, 1-1,5 l sânge este luată, plasma revine) |
| lupta împotriva proliferării mieloide | terapie citostatică (hidroxiuree, interferon-α, anagrelidă (reține supraproducția de trombocite)) |
| tratamentul rezultatelor bolii: | |
| mielofibroza | transfuzii de sânge (eritrocite, masă trombocitară), splenectomie, anabolice |
| leucemie acută | polichimioterapia |
| CML | terapia citostatică |
| tratamentul complicațiilor: | |
| tromboză vasculară | anticoagulante, antiagregante plachetare |
| hipersplenism | îndepărtarea splinei |
| hiperuricemie | alopurinol 300-1000 mg/zi |
BOLI LIMFOPROLIFERATIVE (LIMFOLEUCEMIE CRONICĂ, PLASMOCITOM, LIMFOGRULEMATOZA)
Toate bolile limfoproliferative au o origine comună în celulele sistemului limfatic (adică sistemul imunitar). Acestea includ:
leucemie limfocitară cronică (LLC);
plasmocitom;
limfogranulomatoza (LGM).
Leucemie limfocitară cronică
EPIDEMIOLOGIE
reprezintă 30% din toate leucemiile;
incidenţă: 3-35 la 100.000;
20/100.000 peste 60 de ani;
Imunofenotipul celulelor B se găsește în 96%, celula T - în 2,5%.
În 100% din cazuri sunt detectate aberații cromozomiale, printre care: deleția brațului lung al cromozomului al 13-lea (55%), al 11-lea (18%). Cu leucemie cu celule T - trisomia 7 perechi de cromozomi.
Ganglioni limfatici de mărimea unei nuci, consistență moale, aluoasă, nelipiți între ele, gâtul este netezit.
KLA: leucocitoză, limfocitoză, există prolimfocite, VSH accelerat, umbre Gumprecht, o scădere a nivelului de eritrocite și hemoglobină, neutrofile - doar 1-2%.
Umbrele lui Gumprecht:
semn caracteristic de laborator al LLC;
sunt nucleele distruse ale limfocitelor;
numărul lor nu este un indicator al severității procesului (adică artefact);
nu sunt în sânge lichid, dar sunt pe sticlă (apar când se trece un alt pahar peste acest pahar, în urma căruia limfocitele sunt distruse);
au valoare diagnostică în stadiile incipiente.
Diagnostic diferentiat:
efectuat cu alte boli ale grupului limfoproliferativ, cum ar fi LGM, macroglobulinemia Waldenström (macroglobulina este un pentamer IgM), plasmocitomul;
decide dacă este limfom cu leucemie sau leucemie cu screening.
Complicatii:
susceptibilitate crescută la infecții: un defect al răspunsului imun - o încălcare a interacțiunii limfocitelor T și B;
cu hiperplazie a foliculilor limfatici ai arborelui bronșic și infiltrare de celule tumorale ale țesut pulmonar- atelectazie, încălcarea ventilației și cu adăugarea florei anaerobe - gangrena;
flegmon frecvent (inclusiv din injecții), accesarea infecțiilor nosocomiale;
pleurezie (parapneumonică, tuberculoasă);
tuberculoză (din cauza imunodeficienței);
infiltrarea limfatică a pleurei; cu o ruptură a ductului limfatic - chilotorax;
herpes generalizat (până la moarte);
în stadiul terminal - creșterea sarcomului (hipertermie, diferențiere de TB etc.);
cu infiltrare a parenchimului rinichilor - insuficiență renală cronică;
sindromul de citoliză: hemoliză și anemie, reticulocite în sânge, trombocitopenie până la sindromul hemoragic.
TRATAMENT CLL
terapia citostatică inițială:
cu leucocitoză și LAP moderată: leukeran (clorbutină) 4-10 mg 1 dată pe zi; controlul leucocitozei, dimensiuni l / y; terapie de întreținere: 4-8 mg la două zile - inducerea compensării clinice;
cu leucocitoză moderată și LAP severă: ciclofosfamidă (endoxan) 200-400 mg pe cale orală 1 dată pe zi; terapie intermitentă 200-300-400 mg 1 dată pe zi timp de 10 zile în interior (5 doze), după o pauză de două săptămâni - repetați cursul.
Programe CLL PCT:
CHOP - ciclofosfamidă, vincristină, adriamicină, prednisolon;
COP - ciclofosfamidă, vincristină, prednisolon;
ATS - ciclofosfamidă, adriamicină, prednisolon;
M2 - ciclofosfamidă, carmustină, vincristină, melfalan, prednisolon.
fludarabină (FAMP);
medicamente noi:
gemcitabină;
cladribină (2-clorodesoxiadenozină);
MabThera (rituximab - anticorpi himerici împotriva celulelor B de suprafață CD20);
anticorpi Campath-1H (anti-CD52).
Leucemiile paraproteinemice sunt tumori care secretă Ig monoclonale sau fragmentele acestora, care sunt bine detectate prin electroforeză.
Caracteristici clinice:
sindromul patologiei proteice:
nefropatie (amiloidoză secundară);
polineuropatie;
hipervâscozitatea sângelui până la comă;
încălcări ale hemostazei;
încălcări ale imunității umorale;
sindrom hiperuricemic (cum ar fi guta secundară).
Limfoame secretoare de imunoglobuline:
mielom multiplu;
plasmocitom solitar;
macroglobulinemie Waldenström;
limfoame cu secreție monoclonală de Ig;
boala Ig grea;
dificil de clasificat tumorile secretoare de Ig.
plasmocitom
EPIDEMIOLOGIE
cel mai frecvent dintre cele de mai sus este mielomul multiplu (plasmocitom, boala Rusticki-Kahler) (10-15%);
letalitate = 18%;
A contribui:
predispozitie genetica;
defecte în supresia celulelor T;
influența stimulării antigenice cronice;
afectarea genomului: radiații, chimie (inclusiv medicamente), viruși.
CLINICA
Caracteristicile clinice ale plasmacitomului:
1) Ig monoclonale:
în 70% din cazuri este IgG;
în 20% din cazuri - IgA;
în 5% - lanțuri L.
2) Leziuni osoase și hipercalcemie:
osteoporoza și afectarea osului litic datorită resorbției crescute;
fracturi patologice frecvente, în special ale coloanei vertebrale;
la un nivel de calciu de 2,6-3,5 mmol - o formă ușoară, mai mult de 3,5 mmol - o formă toxică (este necesar să se ingereze mai mult de 3 litri de apă minerală + NaCl + diureză forțată).
3) Tulburări renale:
proteina mielomului (lanțuri ușoare) se acumulează în tubii renali și este reabsorbită de celulele tubilor renali, provocând leziuni;
hipercalcemia poate provoca leziuni renale;
în plasmocitom, amiloidul poate fi prezent provocând leziuni ale glomerulilor.
4) Infecție:
pacienții cu mielom produc puțin anticorpi atunci când sunt provocați cu un antigen, ceea ce duce la un risc crescut de infecții, în special cu bacteriile încapsulate;
umplerea BM cu celule de mielom poate duce la neutropenie;
chimioterapia utilizată pentru tratarea mielomului poate provoca neutropenie.
5) Guta secundara.
6) Polineuropatie.
asimptomatic (lent);
simptomatic:
activ,
remisiuni (1-3 ani),
recidive;
recidiva refractară.
Indicatori de laborator pentru plasmocitom:
proteina totala: crescuta datorita fractiei de γ-globulina (nu atat de crescuta cu proteinurie);
M-gradient în fracția de γ-globuline (proteina monoclonală a mielomului);
VSH foarte mare;
hipercalcemie, creșterea ureei și a creatininei.
TRATAMENT
metoda principală este chimioterapia:
medicamente alchilante: medicamente alkeran, melfalan, sarcolizină, ciclofosfamidă, nitrozuree + combinația lor cu prednisolon;
melfalan + prednisolon (regim M + R) - standard de terapie de primă linie, 6-12 cure → remisiune → terapie de întreținere cu interferon-α în doză de 3 milioane U/m2 de 3 ori pe săptămână, dar recăderile sunt inevitabile; poate fi utilizat melphalan în doze mari;
tratament simptomatic:
fracturi - tratament de către un traumatolog;
bifosfonați pentru osteoporoză;
hidratare în hipercalcemie;
tratamentul insuficienței renale cronice;
plasmafereza (reducerea hipervâscozității și prevenirea comei paraproteinemice);
tratamentul anemiei (masa eritrocitară, dacă cauza anemiei este CRF, atunci eritropoietina);
terapie antibacteriană;
alopurinol (terapia sindromului hiperuricemic).
Limfogranulomatoza
Substratul tumorii: celule Sternberg-Reed (CD23) (retractie citoplasmatica, nuclei mici).
ETIOLOGIE
radiații ionizante;
chimicale: benzen, TNT, insecticide etc.;
medicamente: citostatice, sulfonamide, amidopirină, mercazolil, cloramfenicol etc.;
factori autoimuni;
CLINICA
sindrom circulator-hipoxic (respirație scurtă, tahicardie, slăbiciune, suflu sistolic peste inimă, paloare piele);
sindrom infecțios-toxic (febră, amigdalită, pneumonie, infecții ale tractului urinar, până la dezvoltarea unei stări septice);
sindrom hemoragic (peteșii, echimoze, hematoame, sângerări nazale și uterine);
studiul KM: inhibarea tuturor germenilor hematopoiezei.
leucemie
Concepte generale leucemie. Patogenia generală și cinetica celulară a leucemiilor. Tumori mieloproliferative și limfoproliferative. Leucemie monocitară cronică. Clasificarea hemoblastozelor paraproteinemice. Caracteristici ale morfologiei celulelor leucemice.
Hemoblastozele paraproteinemice (mielomul multiplu, macroglobulinemia Waldenström, boala lanțului greu) sunt tumori ale sistemului limfocitelor B (celule care îndeplinesc funcțiile de imunitate umorală).
Leucemiile sunt numeroase tumori care apar din celulele hematopoietice și afectează măduva osoasă. Durata bolii, etiologie și patogeneză. Cauzele leucemiei acute și cronice, tablou clinic, tratamentul și utilizarea antibioticelor.
Sindromul nefrotic este o afecțiune nefavorabilă din punct de vedere prognostic, un complex de simptome clinice și de laborator. Patogenia edemului în sindromul nefrotic (teoria clasică). Amiloidoza este o boală sistemică bazată pe tulburări metabolice.
În corpul uman nu există doar vase de sânge, ci și așa-numitele vase „albe”. Erau cunoscuți de multă vreme, iar la mijlocul secolului al XVIII-lea cunoștințele despre sistemul limfatic au devenit mai extinse. Din păcate, bolile limfoproliferative nu sunt neobișnuite și pot apărea în orice organ.
sistem limfatic
Joacă un rol destul de important în funcționarea unei persoane: datorită sistemului limfatic, nutrienții sunt transportați, excesul de lichid interstițial este îndepărtat. O altă abilitate importantă este de a oferi imunitate. Lichidul care îndeplinește aceste sarcini se numește limfa. Are o culoare transparentă, compoziția este dominată de limfocite. Cea mai mică unitate structurală a sistemului sunt capilarele. Acestea trec în vase, care sunt atât intraorganice, cât și extraorganice. Structura lor include și supape care împiedică curgerea inversă a fluidului. Cele mai mari se numesc colecționari. În ele se acumulează lichidul din organele interne și din altele mari.O altă componentă pe care o are sistemul limfatic (fotografia este situată mai jos) sunt ganglionii. Acestea sunt formațiuni rotunde care au diametre diferite (de la jumătate de milimetru până la 5 centimetri). Ele sunt situate în grupuri de-a lungul traseului vaselor. Funcția principală este filtrarea limfatică. Aici este curățat de microorganismele dăunătoare.
Organe limfatice
Următoarele organe fac, de asemenea, parte din sistemul limfatic uman: amigdalele, splina, măduva osoasă. Limfocitele care se formează în timus se numesc celule T. Caracteristica lor este circulația continuă între limfă și sânge. Particulele care se formează în măduva osoasă se numesc celule B. Ambele tipuri după maturare sunt răspândite în tot corpul. Celulele B rămân în organele limfoide. Acest lucru le oprește migrația. ÎN cavitate abdominală găzduiește un alt organ mare, care este parte integrantă a sistemului limfatic, este splina. Este format din două părți, una dintre ele (pulpa albă) generează anticorpi. 
Acest grup de boli se bazează pe creșterea limfocitelor. Dacă apar modificări în măduva osoasă, atunci se folosește termenul „leucemie”. Tumorile sistemului limfatic care apar în țesutul din afara măduvei osoase se numesc limfoame. Conform statisticilor, cel mai adesea astfel de boli apar la pacienții mai în vârstă. La bărbați, acest diagnostic apare în Mai mult decât la femei. Această boală este caracterizată de un focar de celule, care în cele din urmă începe să crească. Alocați grad scăzut, mediu și înalt, care caracterizează malignitatea procesului.
Cauze posibile
Printre cauzele care pot provoca boli limfoproliferative se numără un anumit grup de viruși. Factorul eredității joacă, de asemenea, un rol important. Afecțiunile pielii care durează o perioadă semnificativă de timp (cum ar fi psoriazisul) pot determina creșterea neoplasme maligne. Și, desigur, radiațiile afectează în mod semnificativ acest proces. Radiațiile, unii alergeni, substanțele toxice activează procesul de creștere a celulelor.
limfoame. Diagnosticare
Un tip de neoplasm malign al sistemului limfatic este limfomul. Simptomele din stadiile inițiale pot să nu fie foarte pronunțate. 
Există o creștere care nu este dureroasă. Un alt semn izbitor este oboseala, și într-o măsură destul de mare. Pacientul se poate plânge transpirație excesivă noaptea, o pierdere semnificativă și bruscă a greutății corporale. Este posibilă și mâncărimea.Temperatura corpului crește uneori, mai ales seara. Aceste simptome trebuie alertate dacă nu dispar după câteva săptămâni. Pentru tratament eficient Este foarte important să se determine tipul de limfom. Când diagnosticați, luați în considerare locația, aspect tumora, tipul de proteină care se află la suprafața sa. Specialistul prescrie un examen medical complet, un test de sânge pentru celulele canceroase și un diagnostic al organelor interne. Pentru mai multe informații, este necesară o biopsie. La microscop, celulele afectate au un aspect specific.
Tratamentul limfomului
Metode de tratament această boală ca urmare a. Chimioterapia sau radioterapia (folosind raze X) sunt folosite pentru a distruge neoplasmul. Se folosește o combinație de medicamente, acestea sunt distribuite în organism și pot distruge și acele celule care nu au putut fi diagnosticate. După chimioterapie, măduva osoasă este și ea afectată, așa că poate fi necesar să fie transplantată. Se efectuează atât din materialul donator, cât și direct din măduva osoasă a pacientului (este îndepărtat în prealabil înainte de începerea procedurilor). Bolile limfoproliferative sunt, de asemenea, susceptibile de terapie biologică, dar este predominant experimentală. Se bazează pe utilizarea unor substanțe care sunt sintetizate din celulele pacientului. Pentru a obține un rezultat bun, este necesar să urmați cu atenție instrucțiunile medicului curant, să luați medicamentele la timp și să acordați atenția cuvenită nutriției. 
leucemie. Tabloul clinic
Boala se caracterizează printr-o modificare a celulelor hematopoietice, în care elementele sănătoase ale măduvei osoase sunt înlocuite cu cele afectate. Nivelul limfocitelor din sânge crește semnificativ. În funcție de ce celule au renascut, boala este izolat leucemia limfocitară (modificări ale limfocitelor), leucemie mieloidă (mielocitele sunt afectate). Puteți determina tipul de boală la microscop și prin analiza proteinei. Boala limfoproliferativă (ce este, a fost descris mai sus) în acest caz are, desigur, două forme: cronică și acută. Ultima este destul de grea. În acest caz, este necesar un tratament imediat, deoarece celulele sunt imature și nu își pot îndeplini funcțiile. Forma cronică poate dura mulți ani. 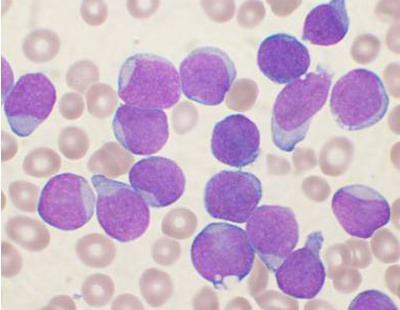
Persoanele în vârstă sunt adesea diagnosticate. Boala decurge destul de lent și numai pentru stadii târzii se observă tulburări în procesul de formare a sângelui. Simptomele includ umflarea ganglionilor limfatici și a splinei, frecvente boli infecțioase, pierdere în greutate, transpirație. Adesea astfel de boli limfoproliferative sunt descoperite întâmplător.
 Există trei etape ale bolii: A, B, C. Primul afectează 1-2 ganglioni limfatici, al doilea - 3 sau mai mulți, dar nu există anemie și trombocitopenie. La a treia se observă aceste stări. Pe primele etape experții nu recomandă terapia, deoarece o persoană își păstrează stilul obișnuit de viață. În același timp, este important să se respecte regimul zilnic, medicul poate da sfaturi cu privire la nutriție. Se efectuează terapie restaurativă. Tratamentul leucemiei limfocitare cronice ar trebui să înceapă atunci când sunt detectate semne de progresie. Include chimioterapie, radioterapie.Odată cu creșterea rapidă a organului, poate fi necesară îndepărtarea splinei.
Există trei etape ale bolii: A, B, C. Primul afectează 1-2 ganglioni limfatici, al doilea - 3 sau mai mulți, dar nu există anemie și trombocitopenie. La a treia se observă aceste stări. Pe primele etape experții nu recomandă terapia, deoarece o persoană își păstrează stilul obișnuit de viață. În același timp, este important să se respecte regimul zilnic, medicul poate da sfaturi cu privire la nutriție. Se efectuează terapie restaurativă. Tratamentul leucemiei limfocitare cronice ar trebui să înceapă atunci când sunt detectate semne de progresie. Include chimioterapie, radioterapie.Odată cu creșterea rapidă a organului, poate fi necesară îndepărtarea splinei.


Grupul de boli limfoproliferative include:
Leucemie limfoblastică acută
Leucemie limfocitară cronică
Limfogranulomatoza (limfom Hodgkin)
limfoame non-Hodgkin (limfosarcoame)






Tabloul clinic al limfoamelor
Simptome de intoxicație
- nu provoacă senzații subiective la pacient și poate fi detectat în timpul unei examinări aleatorii
- slăbiciune, oboseală, febră, scădere în greutate
- transpirații abundente, în special noaptea, mâncărimi nemotivate ale pielii și toleranță slabă la mușcăturile de insecte care suge sânge
Simptomele progresiei tumorii
- Sindromul metastatic (l/a mare)
- Sindrom imunologic (anemie hemolitică imună, trombocitopenie imună, sindrom asemănător lupusului)
- Scăderea stării imunitare (infecții bacteriene și virale frecvente)

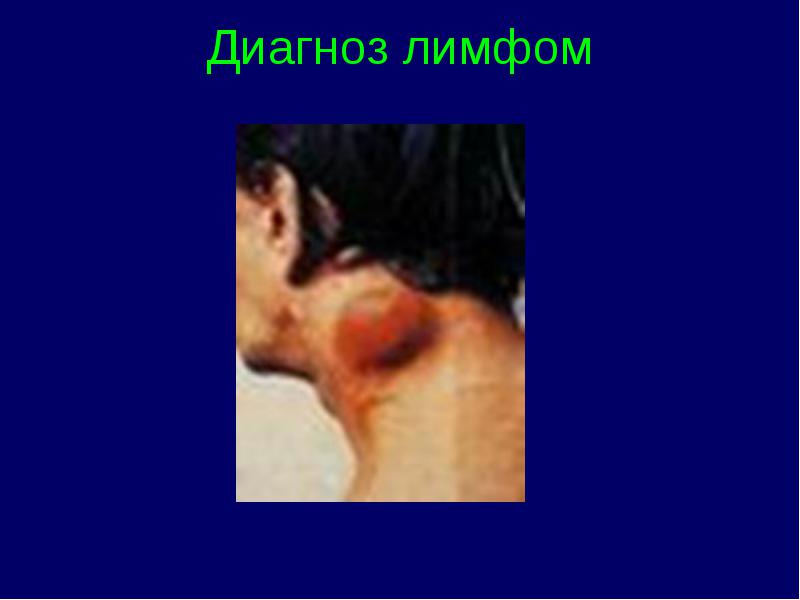
Diagnosticul limfoamelor
Într-un test de sânge:
- Adesea nu există nicio schimbare
- Poate fi anemie, trombocitemie, leucocitoză (limfocitoză, eozinofilie)
- Sindromul citopenic în limfomul MTS din măduva osoasă



Diagnosticul limfoamelor
Diagnosticarea radiațiilor
Metode suplimentare de cercetare(imunofenotiparea prin citometrie în flux, studii citogenetice și genetice moleculare)

Diagnosticul limfoamelor
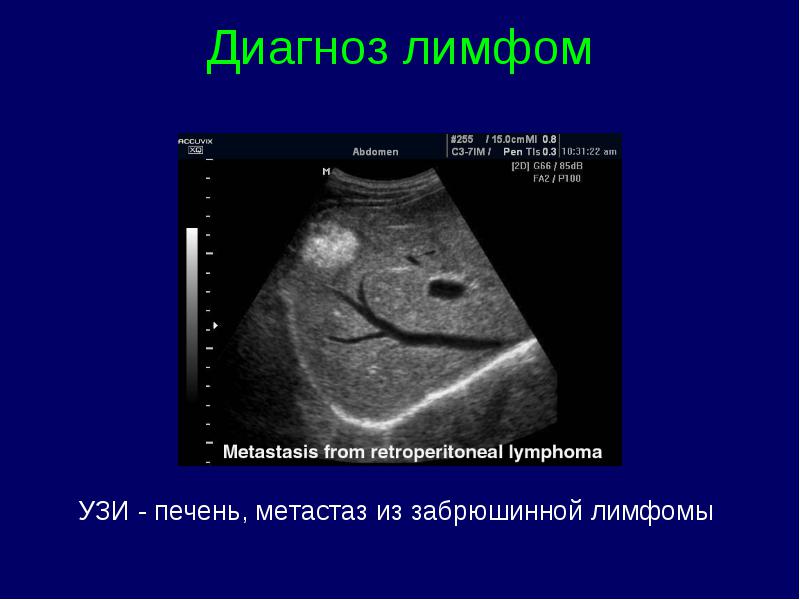
Diagnosticul limfoamelor
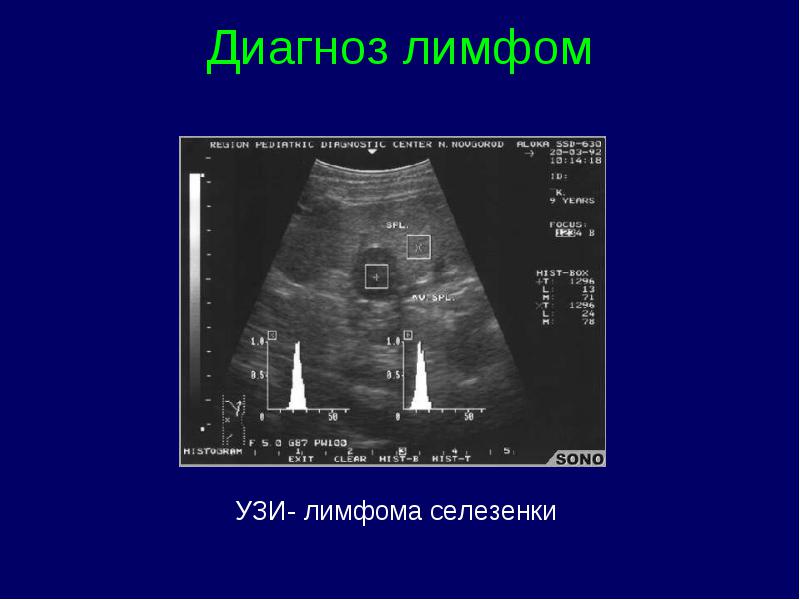
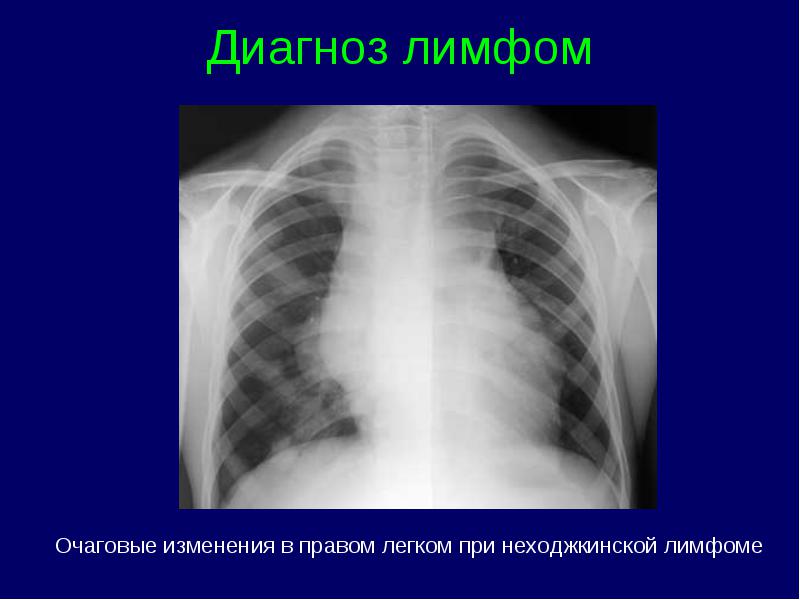
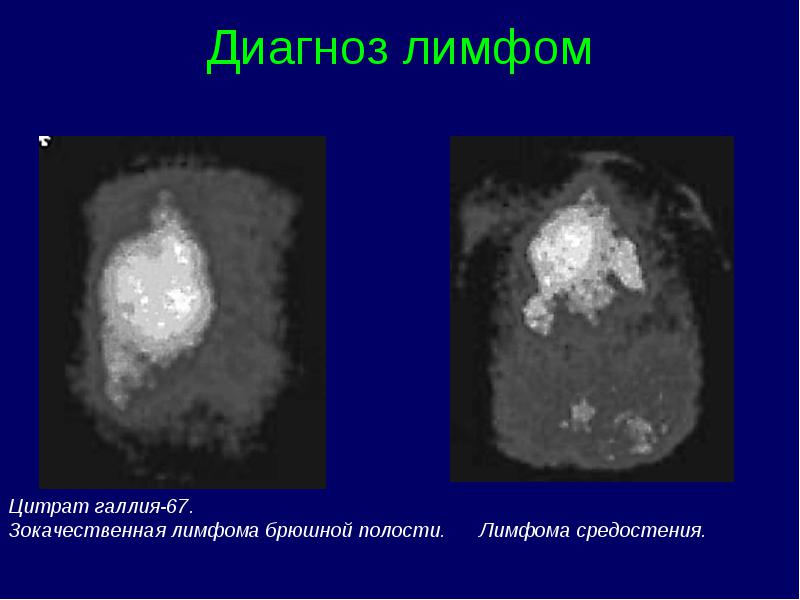
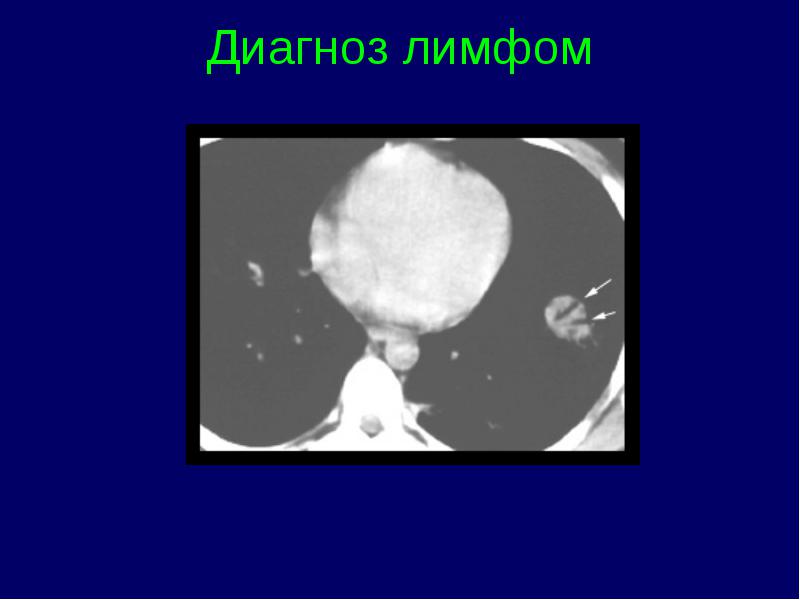

Clasificarea stadiilor limfomului
Simptome de intoxicație care determină substadiile A și B:
transpirații nocturne

O versiune simplificată a diagnosticului limfoamelor non-Hodgkin în funcție de gradul de malignitate:
Limfoamele formate din celule imature (limfoblaste) sunt limfoame de grad înalt.
Limfoame formate din celule de maturitate intermediară (prolimfocite) - limfom de grad intermediar de malignitate.
Limfoamele formate din celule mature (limfocite) sunt limfoame de grad scăzut.

Tratamentul limfoamelor
Polichimioterapia
Chimioterapia în doze mari cu transplant de celule stem hematopoietice.
Terapie cu radiatii


Pacientul P., în vârstă de 72 de ani, a fost internat pentru exacerbarea pneumoniei cronice.
La examinare, se palpează ganglionii limfatici măriți la un ou de găină: cervicali, inghinali, axilari. Ganglionii limfatici sunt moi, nu lipiți între ele
Plămâni: sunet de percuție cu ton de casetă, rale uscate împrăștiate pe ambele părți. Hepatosplenomegalie.
Sânge: Hb - 74 g/l, eritrocite - 2,31012, reticulocite - 20%, leucocite - 5,0109: e - 1%, s/i - 2%, s/i - 17%, limfocite - 79%, monocite - 1%, VSH 60 mm/h, celule leucolice. Trombocitele - 100109.

întrebări interactive
1. Ce boală nu este clasificată ca boală limfoproliferativă:
- Leucemie limfocitară acută
- Leucemie limfocitară cronică
- Hemoblastoze paraproteinemice
- Fibroza măduvei osoase cu hematopoieza extramedulară
- limfomul Burkit

întrebări interactive
2. Care simptom nu definește substadiile A și B:
- creșteri inexplicabile ale temperaturii până la 38 ° C seara cu perioade scurte de afibrilă
- transpirații nocturne
- Pierderea parului
- pierdere în greutate inexplicabilă de peste 10% în șase luni

întrebări interactive
3. Dacă în măduva osoasă punctată sunt detectate celule similare morfologic cu celulele focarului primar al limfomului, ar trebui să se gândească
Transformarea limfomului în leucemie
Metastazele limfomului la măduva osoasă
Dezvoltarea unei crize blastice în limfom

întrebări interactive
4. În ce stadiu al bolii este detectată o creștere a 5 grupe de ganglioni limfatici pe părțile opuse ale diafragmei:

întrebări interactive
5. Ce poate fi în măduva osoasă punctată cu limfomul Hodgkin
- compoziție celulară normală
- limfocitoză până la 20%
- determina celule similare morfologic cu celulele focarului primar al limfoamelor
- Inhibarea tuturor germenilor de hematopoieză
- Determinată de explozii

LITERATURĂ
Curcubeu N.L. Boli interne Mn: VSH, 2007, 365s
Pirogov K.T Boli interne, M: EKSMO, 2005
Sirotko V.L., Totul despre bolile interne: un manual pentru studenții absolvenți, Mn: VSH, 2008

Literatură
Abdulkadyrov K.M. et al. Sindroame hematologice în practica clinică generală // „Elbi”, Sankt Petersburg.-1999.-S.83-94
Sindroame hematologice în practica clinică, editat de Vyagorskaya Ya.I., Kiev „Sănătate” 1981 od
Vorobyov A.I. Ghid de hematologie.// „Newdiamed”, Moscova.-2003.-T1.
Wood M.E., Bunn P.A. Secretele hematologiei și oncologiei// „Binom” - Moscova.-2001.-p.85-93.
Guseva S.A., Voznyuk V.P. Boli ale sistemului sanguin. Director. // „MEDpress-inform”.-Moscova.-2004.-p.317-356.
Boli interne editate de Ryabva S.I., Almazova V.A., Shlyakhto E.V., Sankt Petersburg, SpetsLit, 2000
Oncohematologie clinică, editat de Volkova M.A., Moscova, „Medicina”, 2001
Shiffman F.D. Fiziopatologia sângelui. // Binom.-Moscova.-2000.-p.71-123, 343-358
-
Grup eterogen clinic și morfologic de boli, baza proces patologicîn care apare în principal în piele proliferarea malignă a limfocitelor sunt boli limfoproliferative. Citiți articolul despre cum se dezvoltă bolile limfoproliferative și de ce.
Ce sunt bolile limfoproliferative?
Eterogenitatea limfoamelor maligne a fost stabilită în funcție de tipul de limfocit proliferativ, de apartenența acestuia la o anumită populație și subpopulație.
Distribuția limfocitelor în piele în funcție de fenotip la pacienții cu boli limfoproliferative este aceeași ca și la persoanele sănătoase: limfocitele T sunt localizate în principal în epidermă și straturile superioare ale dermului, iar limfocitele B în straturile mijlocii și profunde ale pielii. dermului. În consecință, tumorile cu celule T ocupă în principal straturile superioare ale dermei, iar procesele limfoproliferative B apar în straturile profunde ale dermului și nu sunt de natură epidermotropă. Numărul de limfoame cu celule T cutanate (TKL) depășește semnificativ numărul de limfoame cu celule B (BCL); Limfoamele T reprezintă 65% din toate limfoamele cutanate maligne, limfoamele B pentru 25% și limfoamele neclasificate pentru 10%.
Cel mai adesea, aceste boli limfoproliferative se dezvoltă la vârstnici, deși există cazuri izolate de boală chiar și la copii. Bărbații suferă de boli limfoproliferative de 2 ori mai des decât femeile.
Cum se dezvoltă bolile limfoproliferative?
Etiologia bolii nu a fost elucidată. La fel de factor etiologic dezvoltarea bolilor limfoproliferative ale pielii, retrovirusuri precum virusul limfotrop uman de tip I (HTVL-I) sunt în prezent luate în considerare, care cauzează leucemie cu celule T la adulți. Dezvoltarea celei mai frecvente forme de boli limfoproliferative ale pielii - micoza fungoide - este asociată cu retrovirusul C, care se găsește la pacienții la nivelul pielii, sânge periferic, celulele Langerhans. Anticorpii împotriva HTVL-I sunt detectați la mulți pacienți cu micoză fungoide.
Rolul factorilor ereditari este posibil. De exemplu, antigenele de histocompatibilitate B-5 și B-35 se găsesc adesea la pacienții cu limfoame cutanate foarte maligne, A-10 în limfoamele mai puțin agresive și B-8 predominant la pacienții cu micoză fungoidă eritrodermică.
dermatoze cronice pe termen lung, cum ar fi neurodermatita, Dermatita atopica, psoriazisul etc., contribuie la persistența pe termen lung a limfocitelor în infiltratele inflamatorii, care, pe fondul influenței factorilor de promovare în condiții de supraveghere imunitară afectată, pot contribui la apariția unei clone de limfocite maligne și, astfel, dezvoltarea unui proces proliferativ malign.
O anumită importanță în geneza bolilor limfoproliferative o joacă radiatii ionizante, radiații UV, diverși compuși chimici cu proprietăți cancerigene și unii alergeni. Acești factori pot duce la apariția unei clone de limfocite „genotraumatice” datorită activării proto-oncogenelor sau inactivării genei supresoare tumorale și, astfel, inițiază procesul de malignizare a limfocitelor.
Cum se dezvoltă bolile limfoproliferative sub influența radiațiilor?
Reacțiile imunopatologice la voluntarii cu simptome de boli limfoproliferative ale pielii sunt efectuate în piele de limfocite și celule ale micromediului: keratinocite și celule Langerhans. Celulele limfocitelor au capacitatea de a recircula continuu în piele. Funcția principală a acestor celule este eliminarea materialului antigenic. Nivelul de recirculare a limfocitelor în timpul stimulării antigenice cronice crește de 10 ori sau mai mult, ceea ce duce la un aflux crescut de limfocite în piele. Pe de o parte, acest proces este util, deoarece limfocitele sunt implicate în reacții protectie imunitara, dar pe de altă parte, crește riscul apariției unei clone mutante de limfocite.
Cum se dezvoltă bolile limfoproliferative - mecanismul dezvoltării bolii
În TCL cutanat, există o proliferare predominantă a limfocitelor T-helper, care au caracteristici funcționale și membranare similare cu limfocitele T-helper sănătoase.
Asta permite perioadă lungă de timpîși îndeplinesc funcțiile imunologice. În plus, membranele acestor limfocite exprimă antigenul limfocitar asociat pielii, care contribuie la afinitatea pe termen lung a celulelor T-helper pentru piele. Cu toate acestea, odată cu creșterea masei clonei tumorale, proprietățile funcționale ale limfocitelor se pierd, în special, antigenul funcțional limfocitar (LFA-1). În același timp, pe fondul bolilor limfoproliferative ale pielii, antigenul nuclear al celulelor în proliferare (Ki-67, un marker al malignității limfocitelor) apare pe membranele celulare ale limfocitelor, iar genele supresoare tumorale (P-53) sunt inactivate. .
Acești factori indică o scădere a supravegherii antitumorale, pierderea afinității, duc la extinderea celulelor tumorale în straturile profunde ale dermei și adesea organe interne. În prezent, a fost identificat un rol deosebit în transformarea tumorii și proliferarea limfocitelor T (celule ale micromediului). celulele Langerhans și keratinocite. Primele sunt o componentă importantă a sistemului macrofagic al pielii, care, la rândul său, face parte din sistemul de supraveghere imunitară. Funcția principală a acestor celule este de a prezenta informații antigenice limfocitelor T. În plus, în bolile limfoproliferative ale pielii, celulele Langerhans sunt capabile să influențeze diferențierea și proliferarea limfocitelor T și să stimuleze generarea de limfocite citotoxice. Aceste funcții ale celulelor Langerhans sunt realizate datorită sintezei de citokine, în primul rând interleukina-6.
Cum se dezvoltă bolile limfoproliferative - simptome histologice
Cu stimulare antigenică prelungită, posibil virală, are loc o scădere a numărului de celule Langerhans și o scădere a utilității lor funcționale, ceea ce perturbă supravegherea imună și, astfel, duce la supraviețuirea și extinderea clonei maligne a limfocitelor T. Se știe că keratinocitele sporesc mult activitatea celulelor Langerhans prin „prezentarea” antigenelor limfocitelor T.
În plus, sunt capabili să sintetizeze citokine, dintre care interleukina 1 (IL-1) este cea mai semnificativă funcțional. S-a stabilit că IL-1 este identică cu factorul de activare a timocitelor epidermice (ETAF) și este capabilă să activeze limfocitele T. Odată cu creșterea producției de această citokină la pacienții cu boli limfoproliferative ale pielii, afluxul de limfocite către leziuni este asociat cu exocitoza ulterioară în epidermă până la formarea de microabcese Potrier în ea, precum și proliferarea limfocitelor în epidermă. dermului. Limfocitele stimulate cu IL-1 sintetizează o altă citokină, IL-2 (factor de creștere a celulelor T), care este de o importanță cheie în patogeneza TCL cutanată. Producția crescută de IL-2 la pacienții cu TKL este, de asemenea, asociată cu înfrângerea presupusului virus limfotrop al subpopulației de limfocite T-helper, ceea ce îi face capabili de creștere „nemuritoare”.
Astfel, procesul de formare a TCL al pielii începe cu activarea limfocitelor sub influența diverșilor factori cancerigeni și apariția unei clone de celule T dominante. Atunci când apare în piele, capacitatea sa de a efectua controlul antitumoral variază într-o mare măsură, determinând dinamica procesului la pacienții cu TCL - de la focare cu plăci neregulate până la dezvoltarea de tumori mari și moarte.
Care sunt tipurile de boli limfoproliferative?
Cea mai comună în Europa este clasificarea Kiel modificată, care ia în considerare parametrii histologici și citologici în bolile limfoproliferative ale pielii. În funcție de dinamica dezvoltării TCL cutanate, de gradul de diferențiere a celulelor tumorale și de micromediu, acestea sunt împărțite în tumori de grad scăzut (I), mediu (II) și înalt (III) de malignitate.
Pentru a diagnostica bolile limfoproliferative ale pielii, este necesar să se evalueze tipul de erupții cutanate (pete, plăci, tumori), rata de apariție a acestora, datele studiilor histologice, citologice, cu raze X și tomografice, rezultatele clinice și parametrii biochimici ai sângelui, starea ganglionilor limfatici. Studiile măduvei osoase sunt justificate numai în cazurile în care se găsesc celule tumorale în noduli limfaticiși/sau în sânge.

