04.01.2018
Prevenirea și tratamentul sindromului hemoragic. Forme dobândite de deficiență a factorilor complexului protrombinic. Simptome și curs
Sindromul hemoragic, sau o tendință la hemoragie cutanată și sângerare a membranelor mucoase, apare ca urmare a modificărilor uneia sau mai multor legături ale hemostazei. Aceasta ar putea fi o înfrângere peretele vascular, încălcarea structurii, funcției și numărului de trombocite, încălcarea hemostazei coagulării. La determinarea cauzelor sângerării, trebuie luat în considerare faptul că unele tipuri de patologie sunt frecvente, altele sunt rare, iar altele sunt extrem de rare. Dintre tulburările ereditare ale hemostazei, trombocitopatiile, hemofilia A, boala von Willebrand, hemofilia B sunt cele mai frecvente în practica terapeutică, iar telangiectazia din formele vasculare. Cele mai frecvente cauze ale formelor dobândite de sindrom hemoragic sunt trombocitopenia secundară și trombocitopatiile, DIC, deficitul de factori ai complexului protrombinic și vasculita hemoragică. Alte forme sunt rare sau foarte rare. Trebuie avut în vedere că în ultimii ani, din ce în ce mai multe tulburări de hemostază și, ca urmare, sindromul hemoragic sunt asociate cu luarea medicamente care încalcă agregarea plachetară (agenți antiplachetari) și coagularea sângelui (anticoagulante), precum și formele psihogene - sângerare nevrotică și sindromul Munchausen.
Caracteristici clinice
Există 5 tipuri de sindrom hemoragic.
- hematom. Tipic hemofiliei A și B, caracterizată prin apariția unor hemoragii intense dureroase la tesuturi moiși articulații, dezvoltarea treptată a disfuncției sistemului musculo-scheletic.
- Petehial-pete (albăstrui). Apare cu trombocitopenie, trombocitopatie, încălcarea sistemului de coagulare (hipo- și disfibrinogenemie, deficiență ereditară a factorilor de coagulare).
- Echimoze mixte-hematom. Se dezvoltă cu o deficiență severă a factorilor complexi de protrombină și a factorului XIII, boala von Willebrand, DIC, o supradoză de anticoagulante și trombolitice, apariția inhibitorilor imunitari ai factorilor VIII și IX în sânge și se caracterizează printr-o combinație de petechiale pete. hemoragii cutanate cu hematoame mari separate în spațiul retroperitoneal, peretele intestinal . Spre deosebire de tipul de hematom, hemoragia în cavitatea articulară apare extrem de rar. Echimozele pot fi extinse și dureroase.
- Vasculita de tip violet. Se observă în vasculita infecțioasă și imună, se transformă ușor în DIC și se caracterizează prin hemoragie sub formă de erupție cutanată sau eritem pe bază inflamatorie, posibilă adăugare de nefrită și sângerare intestinală.
- tip angiomatos. Se dezvoltă în zone de telangiectazii, angioame, șunturi arteriovenoase și se caracterizează prin hemoragii locale persistente asociate cu zone. patologia vasculară.
Cu un anumit grad de probabilitate, este posibilă asumarea patologiei legăturii vascular-trombocite sau de coagulare a hemostazei în funcție de caracteristicile manifestărilor hemoragice.
Semne de afectare a hemostazei vascular-trombocite și coagulare
| semn clinic | modificări de coagulare hemostaza |
Modificări ale vasculare hemostaza trombocitară |
|---|---|---|
| peteșii | Rar | Caracteristică |
| Hematoame disecante | Caracteristică | Rar |
| Echimoza superficiala | Cel mai adesea mare single | De obicei multiplu mic |
| HemartozăHemartroze | Caracteristică | Rar |
| Sângerare întârziată | Ca de obicei | Rar |
| Sângerare de la tăieturi și zgârieturi | Minim | Prelungit, adesea intens |
| Genul pacienților | 80-90% la bărbați | Puțin mai frecvent la femei |
| Indicarea istoricului familial | De multe ori | Rareori |
Pasul inițial în diagnosticul diferențial al sindromului hemoragic este întotdeauna numărul de trombocite. sânge perifericși teste simple de coagulare.
Literatură
- "Ghid boli interne pentru un medic generalist, ed. F. I. Komarov, 2007, ISBN 5-89481-367-0
Legături și surse
Fundația Wikimedia. 2010 .
Vedeți ce este „sindromul hemoragic” în alte dicționare:
SINDROMUL MIELODISPLASTIC- Miere. Sindromul mielodisplazic (SMD) este un grup de afecțiuni patologice caracterizate prin afectarea hematopoiezei la toți germenii, manifestată prin pancitopenie. MDS se transformă adesea în leucemie acută sau anemie aplastică. Frecvență. Crește … Manual de boală
Sindromul hemoragic este o afecțiune în care există sângerare crescută. Se naște din mucoasele nasului, în tract gastrointestinal, în articulații, piele și așa mai departe. Cauzele patologiei pot fi atât defecte genetice, cât și tulburări dobândite.
Tipul de sângerare
Există următoarele tipuri de sângerare:
- hematom.
- Petehial-spotted (microcirculatorii).
- Amestecat.
- Purpuriu vasculitic.
- angiomatos.
Tip hematom
Există hemoragii extinse la nivelul membranei seroase, mușchi, articulații, țesut subcutanat. Ele pot apărea după traumatisme sau intervenții chirurgicale.
petehial-pătat
Tipul microcirculator (sau petehial-spotted) apare în principal pe piele și mucoase. Hemoragiile sunt asimetrice; nu se formează hematoame. Pericolul este hemoragia cerebrală. Tipul petehial-spotted este notat în trombocitopatie și trombocitopenie.
tip mixt
Tipul microcirculator-hematom apare cu combinații de două forme. În același timp, predomină aspectul petehial-pătat. Sângerarea hematomului se observă numai în țesutul subcutanat. Acest tip de sângerare apare cu boala ereditară von Willebrand. Dintre formele dobândite: se observă un tip mixt cu DIC, o lipsă de factori complexi de protrombină, o supradoză de trombolitice și anticoagulante.
Tipul purpuriu vasculitic
Apare o erupție cutanată sau eritem (roșeață). Sindromul hemoragic se manifestă la nivelul extremităților inferioare. Papule, vezicule, vezicule sunt situate simetric în zona articulațiilor mari. Cea mai frecventă boală din acest grup este Shenlein-Genocha. Vasculita hemoragică se transformă în DIC. La aceasta se pot adăuga sângerări intestinale și nefrită.
Tip angiomatos
Pete mici luminoase, papule ies deasupra suprafeței pielii. Sunt localizate pe piele și mucoase. Când sunt apăsate, petele se estompează. Acest tip de sângerare se observă cu angioame, telangiectazie, șunturi arteriovenoase.
Când se pune un diagnostic, este important să se determine tipul de sângerare. Patologia poate fi asumată de trăsăturile manifestărilor hemoragice. În funcție de semnele existente, se determină în ce legătură de hemostază s-au produs modificările.
Tulburări ale sistemului trombocitar
Trombocitopenia apare atunci când compoziția cantitativă se modifică. La tulburari functionale se dezvoltă o boală precum trombocitopatia.
Trombocitopenie
Această boală se dezvoltă atunci când nivelul trombocitelor este sub normal. Producția de trombocite în măduva osoasă este redusă atunci când funcția splinei este afectată. Cu această patologie, sângerarea este adesea asociată cu tăieturi, nazale, după extracția dentară, menstruale, de origine neclară.
Boala poate fi atât ereditară, cât și dobândită. ÎN vârstă fragedă copiii se îmbolnăvesc la fel, indiferent de sex. Se observă că cel mai adesea boala afectează pacienții tineri cu vârsta cuprinsă între 2 și 8 ani. Femeile se îmbolnăvesc de 2 ori mai des decât bărbații. Cel mai mult, ei sunt sensibili la această boală între 20 și 50 de ani.
Etiologie
Diverse infecții contribuie la dezvoltare purpură idiopatică. La copii, boala se dezvoltă după SARS și infecții precum rubeola, rujeola, varicelă. Insolația excesivă a pielii poate provoca, de asemenea, boala. Sindrom hemoragic observat după administrare preparate medicale care perturbă agregarea trombocitelor.
Mai des, trombocitopenia apare din cauza:
- diverse infecții și intoxicații;
- reacție alergică la medicamente;
- imunitate slabă;
- deficit de B12 și acid folic;
- transfuzii de sânge (dacă grupurile nu se potrivesc);
- leucemie;
- hepatită și insuficiență renală;
- lupus.
Trombocitopenia indusă de medicamente este cauzată de:
- antibiotice și sulfonamide;
- analgezice;
- diuretice;
- sedative.
Simptome și curs
Simptomul principal este sângerarea cavității bucale, a mucoaselor gingiilor, a nasului. Femeile suferă de menstruații abundente, care se pot transforma în sângerări uterine. trăsătură caracteristică trombocitopenia sunt peteșii. Hemoragiile intradermice se pot forma chiar și dintr-o lovitură ușoară. Sunt situate pe suprafața frontală a corpului și pe membre.
Cu purpura trombocitopenică, există sângerări pulmonare, renale și gastrointestinale. Dacă pierderea de sânge este prelungită și frecventă, un astfel de pacient devine invalid. Pacientul are ameteli si slăbiciune generală. Acest lucru se datorează dezvoltării anemiei; in acelasi timp scade cantitatea de hemoglobina si globule rosii.
Tratament
Când sângerează, pacientului i se recomandă repaus la pat. Se prescriu infuzii cu clorura de calciu, transfuzii de sange, vitaminele K si C. doze mari i se administrează imunoglobuline. O metodă radicală pentru progresia bolii este splenectomia (înlăturarea splinei). Dacă după administrare apare sindromul hemoragic medicament, este necesar să anulați acest medicament.
Trombocitopatie
Boala apare atunci când există o încălcare a hemostazei, care este cauzată de disfuncția trombocitelor și inferioritatea lor calitativă. Trombocitopatiile sunt congenitale și dobândite.
Clasificare
Boli trombocite ereditare:
- sindromul Bernard-Soulier;
- trombastenia Glanzman, anomalia May-Hegglin;
- patologii complexe și disfuncții ale trombocitelor;
- boli în care aderența trombocitelor este afectată;
- boli cauzate de probleme cu granulele.
Formele dobândite sunt observate la administrarea medicamentelor și o lipsă de vitamina C. Trombocitopatia apare și cu următoarele patologii:
- DIC;
- tumori;
- anemie asociată cu deficit de cianocobalamină;
- ciroză.
Simptome
Principalele simptome sunt sângerări de la mucoase, gastrointestinale și uterine. Boala poate provoca:
- infecții;
- trauma;
- boli cronice ale ficatului, rinichilor, glanda tiroidași sistemele de sânge
- expunere la soare;
- operațiuni;
- medicamente;
- UHF și UVI (fizioterapie).
Tratament
Pentru tratament se folosesc medicamente care cresc coagularea sângelui. O atenție deosebită se acordă măsurilor preventive. Aceasta include tratamentul bolii de bază, terapia cu vitamine, angioprotectorii, inhibitorii fibrinolizei, medicamentele metabolice. În același timp, procedurile care pot provoca o boală nu sunt prescrise. Se acordă atenție alimentatie buna bolnav.

coagulopatie
Tulburările de coagulare a sângelui pot fi congenitale sau dobândite. Se disting următoarele boli congenitale:
- hemofilie;
- boala von Willebrand.
Hemofilia este boala ereditara care lovește bărbații. Femeile sunt purtătoare ale bolii.
Etiologie și patogeneză
Hemofilia apare atunci când nu există o sinteză insuficientă sau o anomalie a unuia dintre cei trei factori antihemofili. Genele care cauzează hemofilia A și B sunt localizate pe cromozomul X. Așa se explică faptul că boala se înregistrează doar la băieți. Hemofilia A este cauzată de un deficit de factor VIII. Boala de grup B (deficit de factor IX) nu se distinge clinic, dar este tratată diferit.
Simptome
Sângerarea apare după tăieturi și răni minore, vânătăi, extracție dentară. Ele pot apărea chiar și la câteva ore după leziune. Hemoragiile pot fi în regiunea abdominală și articulații. Hematoamele apărute comprimă vasele. Uneori, sângele apare în urină.
Tratament
În hemofilia A, se folosește crioprecipitatul - un preparat de factor VIII. Pentru tratamentul hemofiliei B se folosește plasmă proaspătă congelată, concentrat de factor IX. Vitamina K poate fi folosita ca agent hemostatic.Pentru a preveni dezvoltarea hemartrozei se foloseste un remediu precum Hidrocortizonul. Preparatele cu factor VIII sunt utilizate în boala von Willebrand.
Este necesar să se protejeze pacientul de orice răni și tăieturi. Trebuie să existe mâncare în dieta lui, bogat în vitamine DIN.
DIC
Această boală este observată cu o deficiență a sistemului de coagulare. Acest sindrom hemoragic se caracterizează prin sângerare crescută, o erupție cutanată petechială și disfuncție de organ. DIC se poate dezvolta rapid sau cronic.
Simptome
Manifestările clinice variază în funcție de rata de dezvoltare a bolii. Dacă se dezvoltă cu viteza fulgerului, apar simptome periculoase:
- stare de șoc (pierderea cunoștinței, hipotensiune arterială);
- edem pulmonar;
- insuficiență respiratorie acută;
- distrofie miocardică.
Un curs fulminant este caracteristic infecțiilor stafilococice și anaerobe, mușcături de șarpe, embolie a lichidului amniotic.

În forma subacută, apar erupții cutanate, hematoame și vânătăi, sângerări. Pielea este palidă și rece la atingere. Se observă un curs subacut cu leucemie, pneumonie, otrăvire cu acid acetic.
Forma cronică de DIC poate fi asimptomatică. Cu această boală, rănile nu se vindecă bine. Un alt moment enervant infecții purulente. curs cronic observat la boli oncologice, ciroză hepatică, insuficiență cardiacă.
Tratament
La curs acut pacienții sunt internați, după care se efectuează terapia anti-șoc. Mare importanță are un diagnostic precoce. Cu complicații septice, se administrează antibiotice.
Pentru tratamentul DIC se folosesc anticoagulante, agenți antiplachetari, fibrinolitice și corticosteroizi. Când boala se administrează heparină și plasmă proaspătă congelată. Pentru hemoragiile cutanate se aplică un burete hemostatic.
1. RELEVANȚA TEMEI
Bolile și sindroamele hemoragice reprezintă un grup eterogen de boli din punct de vedere al etiologiei, patogenezei și evoluției. Manifestare generală pentru întregul grup - o tendință la creșterea sângerării.
Cunoașterea și înțelegerea corectă a patogenezei și manifestărilor clinice ale bolilor hemoragice va permite o căutare diagnostică în direcția corectă și alegerea terapiei adecvate în fiecare caz în parte.
2. SCOPUL LECȚIEI
Să fie capabil să suspecteze și să justifice un program de management al pacienților cu sindrom hemoragic bazat pe cunoașterea etiologiei, patogenezei, manifestărilor clinice, metodelor de diagnostic și principiilor de terapie pentru diverse boli hemoragice.
3. ÎNTREBĂRI DE PREGĂTIRE PENTRU LECȚIE
1. Conceptul de „boli și sindroame hemoragice”, definiție.
2. Clasificarea bolilor și sindroamelor hemoragice.
3. Patogenia bolilor și sindroamelor hemoragice.
4. Manifestări clinice ale bolilor și sindroamelor hemoragice.
5. Metode de diagnosticare a bolilor și sindroamelor hemoragice. Diagnosticul screening al tulburărilor de hemostază.
6. Principii de bază ale tratamentului pacienţilor cu boli şi sindroame hemoragice.
7. Evaluarea prognosticului pacienţilor cu boli şi sindroame hemoragice.
4. TESTARE LA NIVEL DE BAZĂ
1. Numiți testele de diagnostic incluse în diagnosticul screening al tulburărilor de hemostază:
A. Determinarea numărului de trombocite din sânge. B. Determinarea timpului de sângerare.
b. cuantificarea factori de coagulare ai plasmei.
D. Definiţia APTT.
D. Toate testele de mai sus.
2. Selectați testele de diagnostic utilizate pentru a detecta tulburările în legătura de coagulare a hemostazei:
3. Selectați testele de diagnostic utilizate pentru a detecta încălcări ale legăturii hemostazei trombocitelor:
A. Determinarea numărului de trombocite din sânge. B. Evaluarea proprietăților funcționale ale trombocitelor.
B. Determinarea timpului de sângerare.
D. Determinarea timpului de protrombină. D. Definiţia APTT.
4. Selectați testele de diagnostic utilizate pentru a detecta tulburările în legătura trombocită-hemostază vasculară:
A. Determinarea timpului de protrombină. B. Definiția APTT.
B. Determinarea timpului de coagulare a sângelui. G. Testul manșetei.
5. Selectați clinic și semne de laborator caracteristica hemofiliei:
A. Bărbații se îmbolnăvesc.
B. Femelele nu fac niciodată hemofilie.
B. Timpul de coagulare a sângelui este prelungit. D. Timpul de sângerare este prelungit.
D. Numărul de trombocite este în limitele normale.
6. Selectați semnele clinice și de laborator caracteristice trombocitopeniei:
A. APTT este prelungit.
B. Timpul de sângerare este prelungit.
B. Hemoragiile la nivelul articulațiilor sunt tipice. G. Doar bărbații se îmbolnăvesc.
D. Unul dintre simptome clinice sunt menoragii.
7. Selectați semnele clinice și de laborator caracteristice bolii Rendu-Osler:
A. Cazurile familiale sunt frecvente. B. Timpul de sângerare este prelungit.
B. Sângerări recurente la una sau două localizări. D. Hemoragii la nivelul articulațiilor.
D. Hemoragii frecvente la nivelul creierului.
8. Selectați semnele clinice și de laborator tipice pentru vasculita hemoragică:
A. APTT în limite normale.
B. Timp de sângerare în intervalul normal.
B. Hemoragii la nivelul articulațiilor mari.
D. Sângerări recurente din nas. D. Niciuna dintre cele de mai sus.
9. Selectați medicamentele indicate pentru tratamentul vasculitei hemoragice:
A. Vitamina K (Vikasol*). B. Heparina.
B. Preparate de calciu. D. Corticosteroizi.
D. Toate medicamentele de mai sus.
10. Selectați medicamentele indicate pentru tratamentul purpurei trombocitopenice idiopatice:
A. Vitamina K (Vikasol*).
B. Antihistaminice.
D. Corticosteroizi. D. Citostatice.
5. PRINCIPALE ÎNTREBĂRI ALE TEMEI
Bolile și sindroamele hemoragice reprezintă un grup de boli și afecțiuni ereditare și dobândite, a căror caracteristică clinică principală este creșterea sângerării sau tendința organismului de a resângera și hemoragia care apare spontan sau după leziuni minore.
În mod normal, hemostaza este realizată cu participarea a 3 componente funcționale și structurale interdependente:
perete vase de sânge(în primul rând - intimitate);
Celule sanguine (trombocite);
Sisteme enzimatice plasmatice (coagulare, fibrinolitică etc.).
Căptușeala interioară a vaselor de sânge și a trombocitelor sunt în mod deosebit strâns legate. Adesea ele sunt combinate într-un mecanism comun de hemostază vascular-trombocitară. Este denumită și hemostază primară, deoarece microvasele și trombocitele joacă un rol principal în oprirea inițială a sângerării. Formarea cheagurilor de coagulare (fibrină) are loc ceva mai târziu și, prin urmare, hemostaza de coagulare poate fi desemnată ca secundară.
5.1. Etiologie și patogeneză
Toate bolile hemoragice pot fi ereditare sau dobândite în natură și pot fi asociate cu patologia uneia sau mai multor componente ale sistemului hemostază.
5.1.1 Clasificarea bolilor și sindroamelor hemoragice
Clasificarea se bazează pe încălcări în diferite părți ale sistemului de hemostază.
Bolile cauzate de tulburări ale hemostazei coagulării se numesc coagulopatie:
Determinate genetic: hemofilia A și B, boala von Willebrand, deficiența oricăruia dintre factorii de coagulare a sângelui;
Dobândite: boli cauzate de o deficiență a factorilor plasmatici de sânge de motive externe: deficit de vitamina K, boli hepatice, apariția de anticorpi specifici la factorii individuali de coagulare a sângelui; fibrinoliza intensificată cauzată, printre altele, de tratamentul cu anticoagulante directe și indirecte, fibrinolitice.
Bolile cauzate de o încălcare a legăturii celulare a hemostazei sunt numite trombocitopenii și trombocitopatii.
trombocitopenie- acesta este un grup de boli (deseori dobândite), în care numărul de trombocite este determinat sub norma existentă.
Trombocitopenia se poate dezvolta din cauza:
Formarea insuficientă a trombocitelor în măduva osoasă (inhibarea proliferării trombocitelor în leziuni chimice, radiații, tumorale);
Creșterea consumului de trombocite (tromboză, DIC etc.);
Distrugerea crescută a trombocitelor (imunitar cu participarea anticorpilor, hemangiom mecanic etc.).
În patogeneza trombocitopeniei, cel din urmă mecanism este cel mai frecvent.
Trombocitopatii sunt caracterizate prin afectarea funcției trombocitelor (ereditare sau dobândite) cu un conținut cantitativ normal de trombocite în sânge. Principalele funcții ale trombocitelor sunt agregarea (lipirea celulelor împreună) și aderența (lipirea trombocitelor de endoteliul vascular). Încălcarea agregării și (sau) aderenței este unul dintre mecanismele de formare a defecte ale hemostazei trombocitelor (primare).
Pentru boli asociate cu tulburări în legătura vasculară a hemostazei, raporta:
Modificări patologice determinate genetic în peretele vascular - telangiectazie hemoragică ereditară a lui Randu-Osler;
Leziuni dobândite ale vaselor de sânge de etiologie imunocomplex - vasculită hemoragică(boala Schönlein-Henoch).
5.2. Manifestări clinice ale bolilor hemoragice
5.2.1. Hemofilia A și B
Boala se transmite printr-un model de moștenire recesiv legat de X, care duce la o producție insuficientă sau o anomalie structurală a factorului VIII (hemofilie A) sau factorului IX (hemofilie B) la bărbați.
Hemofilia A reprezintă 68-78% din coagulopatii, iar hemofilia B - 6-13%.
Încălcarea legăturii de coagulare a hemostazei duce la sângerare tip hematom - hemoragii masive, profunde, dureroase, intense sunt observate în articulațiile mari, mușchi, țesuturi subcutanate și retroperitoneale, sub aponevroză și fascie. Hemoragiile de hematom pot fi spontane, posttraumatice, postoperatorii, adesea tardive, i.e. care apar la câteva ore după accidentare sau intervenție chirurgicală.
Manifestările clinice se caracterizează prin evoluția vârstei:
La naștere - cefalohematoame, hemoragii subcutanate și intradermice, sângerare tardivă din cordonul ombilical;
Perioada de la 1 la 5 ani - sângerare extinsă de la gingii (dentiție), hematoame traumatice (vânătăi, căderi, răni cauzate de obiecte de uz casnic, jucării etc.);
5 ani și mai mult - hematoame în articulațiile mari, deseori hemoragie renală recurentă, sângerare din tractul gastrointestinal.
Complicații ale hemofiliei:
Complicații auto și izoimune asociate cu o creștere a titrului de anticorpi-inhibitori ai factorilor de coagulare VIII și IX (de la 5 la 15%);
Posibil hepatita B;
Sindromul reumatoid secundar, de obicei la 25-30 de ani - un proces inflamator în articulațiile simetrice ale mâinilor și picioarelor fără hemartroză anterioară;
Hematoame intermusculare extinse cu injecții intramusculare - pentru pacienții cu hemofilie, toate medicamentele sunt administrate numai intravenos.
5.2.2. Purpura trombocitopenică idiopatică (autoimună).
Se caracterizează printr-o scădere a trombocitelor sub 150.000 datorită unei scurtări accentuate a speranței de viață a acestora la câteva zile și ore (într-un ritm de 7-10 zile), cauzată de acțiunea autoanticorpilor.
Manifestarea clinică a trombocitopeniei (de obicei, cu această boală, numărul de trombocite este de 50.000 sau mai puțin) este dezvoltarea sângerării de tip petechial-spotted (capilar). Erupții petehiale și vânătăi apar pe pielea extremităților, suprafața anterioară a trunchiului, mai rar pe față, mucoasele buzelor,
cavitatea bucală, conjunctiva ochilor, precum și sângerări caracteristice din mucoasele (gingii, căi nazale), mai rar renale, uterine, din tractul gastro-intestinal. Sângerarea începe spontan, hemoragiile se dezvoltă mai des din cauza vânătăilor, chiar și a celor minime. Sângerarea recurentă după încetarea acesteia este mai puțin frecventă. Ficatul și splina nu sunt mărite. Complicatii:
Scăderea rezistenței capilare, deoarece în trombocitopenie se observă disfuncția endoteliului vascular;
Cu pierderi semnificative de sânge - anemie acută posthemoragică, cu un curs lung al bolii - anemie cronică cu deficit de fier.
5.2.3. Trombocitopatie
Boala lui Von Willebrand se caracterizează atât prin încălcarea sistemului de coagulare a hemostazei (defect de factor VIII), cât și prin încălcarea legăturii celulare a hemostazei (încălcarea aderenței trombocitelor cu numărul lor normal). Defectul factorului VIII este moștenit autozomal. Atât bărbații, cât și femeile sunt afectați. Un tip mixt de sângerare capilar-hematom este caracteristic: hemoragii petechiale și vânătăi, rar - o cantitate mică de hemoragie în articulații (nu duc la deformări) și țesutul retroperitoneal. În cazuri severe - sângerare spontană și post-traumatică din cavitatea bucalăși tractul gastrointestinal, la femei - sângerare uterină.
5.2.4. Vasculita hemoragica (boala Schoenlein-Henoch, vasculita imunocomplex)
Una dintre cele mai frecvente boli hemoragice, care se bazează pe leziunea IgA-imunocomplex a vaselor mici cu dezvoltarea trombozei și hemoragiei. Această leziune a legăturii vasculare a hemostazei corespunde tipului de sângerare vasculitic-violet. Se manifestă ca o erupție papulară cu mâncărime cu mici puncte (de la 2 la 5 mm) (papulele ies deasupra suprafeței pielii, sunt bine palpabile și nu dispar la presiune). Ulterior apar vezicule cu continut hemoragic.
Erupțiile hemoragice sunt simetrice și localizate pe membre, articulații afectate, fese. De obicei erupția este monomorfă și dispare după 2-3 zile, lăsând pigmentarea. În cazul erupțiilor cutanate repetate, erupția este polimorfă.
În aval emit o formă fulgerătoare (de la câteva ore la câteva zile), formă ascuțită(până la 6 luni), formă cronică (mai mult de 6 luni cu exacerbări și remisiuni pe termen lung).
Vasele de orice localizare pot fi implicate în procesul patologic, dar vasele pielii, intestinelor și rinichilor suferă cel mai adesea. În proces sunt implicate și articulațiile mari.
ÎN tablou clinic Se obișnuiește să se distingă patru sindroame:
1) piele;
2) articular;
3) abdominale;
4) renale.
Combinația dintre sindroamele 1 și 2 se numește simplă.
Toate sindroamele pot fi combinate între ele în opțiuni diferite, iar în caz de recidivă, boala poate manifesta fie aceleași, fie sindroame diferite.
Sindromul pielii observată la toți pacienții - aceasta este o erupție cutanată hemoragică papulară, ale cărei caracteristici au fost descrise mai devreme.
Sindromul articular caracterizată prin leziuni simetrice ale articulațiilor mari, de obicei extremitati mai joase. Sunt posibile atât artralgia, cât și artrita tipică. Adesea, artrita este însoțită de mialgie și umflarea extremităților inferioare. Procesul inflamator durează 1-2 săptămâni, nu lasă deformări.
Sindromul abdominal apare cel mai adesea în copilărie și bătrânețe. Creșterea permeabilității peretelui vascular mezenteric duce la impregnarea hemoragică a peretelui intestinal, care provoacă dureri abdominale intense brusc, diaree, greață, vărsături și se complică cu sângerare gastrointestinală în 50%. Sângerarea abundentă este însoțită de colaps și anemie posthemoragică. Adesea cu sindrom abdominal observati febra, leucocitoza si intervale nedureroase (1-3 ore).
sindrom renal decurge după tipul de acută sau glomerulonefrita cronică cu micro-macrohematurie, micro-macroproteinurie. Sindromul nefrotic este rar hipertensiune arteriala necaracteristic. În cursul cronic al bolii și exacerbările frecvente, este posibilă formarea CRF.
Sindromul pulmonar. Vasele pulmonare sunt afectate extrem de rar, iar hemoragiile pulmonare apar și mai rar.
sindrom cerebral foarte rare: dureri de cap, hematoame subdurale, accidente vasculare cerebrale hemoragice.
5.2.5. Telangiectazie hemoragică ereditară (boala Rendu-Osler, angiomatoză hemoragică)
Boală ereditară, transmisă în mod autosomal dominant. În această boală, insuficiența dezvoltării subendoteliului și conținutul scăzut de colagen din acesta în anumite zone pat vascular duce la subțierea focală a pereților microvaselor, extinderea acestora și formarea de angioame cu pereți subțiri. Uneori, șunturile arteriovenoase se formează în plămâni și în alte organe. Telangiectazie devreme copilărie nu sunt vizibile și încep să se formeze abia la vârsta de 6-10 ani, cel mai adesea pe zona de tranziție a buzelor și pe aripile nasului. Ulterior, telangiectaziile apar pe obraji, sprâncene, limbă, mucoasa nazală și bucală, precum și pe alte zone ale pielii: scalp, vârful degetelor, sub unghii. Telangiectaziile se pot forma pe membranele mucoase ale faringelui, laringelui, bronhiilor, în tot tractul gastrointestinal, în pelvisul renal și tractului urinar, în vagin.
În boala Randu-Osler se observă un tip de sângerare angiomatoasă. Se caracterizează prin sângerare recurentă persistentă 1, 2, rareori 3 localizări. Nu există hemoragii spontane sau post-traumatice. În cele mai multe cazuri, manifestările hemoragice ale bolii Randu-Osler încep cu sângerări recurente.
Contribuie la creșterea sângerării boli inflamatorii membranele mucoase (de exemplu, rinita), leziunile lor mecanice (chiar ușoare), situații stresante, efort fizic, luând alcool, alimente picante și murate (provoacă o încălcare a agregării trombocitelor), medicamente, care includ acid acetilsalicilic, AINS.
La unii pacienți cu angiomatoză hemoragică, inferioritatea țesutului conjunctiv se manifestă prin extensibilitate crescută a pielii, slăbiciune aparatul ligamentar(luxații obișnuite), prolaps de valvă mitrală.
Complicațiile includ:
Dezvoltarea anemiei posthemoragice;
Sângerare letală din telangiectazii la plămâni, tractul gastrointestinal, tractul urinar (rar);
Formarea șunturilor arteriovenoase cu dezvoltarea dificultății de respirație, cianoză, eritrocitoză secundară.
5.3. Diagnosticare
Repere clinice importante pentru diagnosticul bolilor hemoragice.
1. Date anamnestice privind durata bolii, posibila geneza ereditară a acesteia, sexul bolnavilor.
2. Date privind bolile de fond și anterioare, precum și efectele exogene, care pot fi asociate cu sângerări patologice.
3. Determinarea tipului de sângerare (pe baza luării în considerare a tuturor fenomenelor de sângerare observate) la pacient și rudele acestuia.
Metodele de cercetare de laborator includ următoarele grupuri:
Studiul legăturii de coagulare a hemostazei;
Determinarea conținutului de fibrinogen și produși de degradare a fibrinei;
Studiul legăturii trombocitelor a hemostazei;
Studiul legăturii microcirculatorii-vasculare a hemostazei;
Metode imunochimice și radioimunochimice pentru studiul hemostazei.
Prima grupă include următoarele teste:
timpul de coagulare a sângelui total (WBC);
Determinarea timpului de protrombină;
Determinarea timpului de trombină;
Definiţia APTT.
Al doilea grup include toate metodele pentru determinarea cantitativă a fibrinogenului, studiul conținutului de produse de fibrinoliză (PDF).
Al treilea grup include următoarele teste:
Determinarea numărului de trombocite, studiul morfologic al acestora;
Determinarea funcției trombocitelor: aderență, agregare;
Determinarea timpului de sângerare.
Al patrulea grup include studii de rezistență capilară, teste de manșetă, cupping, simptome de ciupire, garou etc.
Testele imuno- și radiochimice includ toate studiile cu anticorpi specifici împotriva factorilor individuali de coagulare.
Diagnosticul screening al tulburărilor de hemostază:
timpul de sângerare;
Numărul de trombocite;
timpul de coagulare a sângelui;
5.4. Tratamentul bolilor hemoragice
5.4.1. Hemofilie
1. Terapie de substituție:
concentrat de factor VIII sau IX;
Crioprecipitat de factor VIII sau IX;
Plasmă proaspătă congelată.
2. Plasmafereza (pentru a elimina anticorpii la factorii de coagulare).
3. Luarea inhibitorilor de fibrinoliză (aminocaproic, aminometilbenzoic, acid transesxamic).
4. Terapia hemostatică locală (bureți hemostatici).
5. Preparatele de calciu și Vikasol * nu sunt afișate.
6. Tratamentul complicațiilor:
Anemia feripriva - preparate cu fier;
Hemartroza - puncția articulațiilor și introducerea hidrocortizonului. Toți pacienții cu hemofilie sunt înregistrați în dispensar.
5.4.2. Purpura trombocitopenică
corticosteroizi;
splenectomie;
Imunosupresoare;
Cu sângerare - inhibitori ai fibrinolizei. Contraindicat Produse alimentare(alcool, cofeină, oțet) și
medicamente (AINS) care reduc agregarea trombocitelor.
Toți pacienții sunt înregistrați în dispensar.
5.4.3. Vasculita hemoragică
Spitalizare;
Dieta hipoalergenică;
AINS (sindrom simplu);
corticosteroizi;
heparină (sindrom abdominal);
Citostatice (sindrom renal);
Plasmafereza.
5.4.4. boala Rendu-Osler
Terapia hemostatică locală și generală.
6. CURAREA PACIENȚILOR
Sarcini de supraveghere:
Formarea abilităților de interogare și examinare a pacienților cu boală hemoragică;
Formarea abilității de a face un diagnostic preliminar al bolii hemoragice pe baza datelor anchetei și examinării pacientului;
Formarea abilității de a elabora un program pentru examinarea unui pacient cu diateză hemoragică;
Formarea abilității de a elabora un plan de tratament pentru un pacient cu diateză hemoragică.
Cura pacientilor este muncă independentă elevi. Elevii personal sau într-un grup de 2-3 persoane efectuează un sondaj, examinări, discuții despre starea pacientului, formează un studiu preliminar și diagnostic clinic, alcătuiți un plan de examinare, tratament, determinați prognosticul bolii.
Elevii raportează rezultatele lucrării întregului grup, le discută colectiv.
7. ANALIZA CLINICĂ A PACIENTULUI
Sarcinile analizei clinice:
Demonstrarea metodelor de examinare și chestionare a pacienților cu boli hemoragice;
Controlul abilităților studenților de examinare și chestionare a pacienților cu boli hemoragice;
Demonstrarea metodologiei de realizare a unui diagnostic preliminar al bolii hemoragice pe baza datelor anchetei și examinării pacienților;
Demonstrarea metodologiei de întocmire a unui plan de examinare a pacienţilor cu boli hemoragice;
Demonstrarea metodologiei de formulare a diagnosticului de boală hemoragică;
Demonstrarea metodei de întocmire a unui plan de tratament.
Analiza clinică este efectuată de un profesor sau studenți sub supravegherea directă a unui profesor. Pe parcursul lecției sunt analizate cele mai tipice și/sau complexe situații clinice din punct de vedere diagnostic și/sau terapeutic.
La sfârșitul analizei, trebuie formulat un diagnostic clinic final (dacă nu este posibil, un diagnostic clinic preliminar) și trebuie întocmit un plan pentru examinarea și tratamentul unui pacient cu o boală hemoragică.
8. SARCINI SITUAȚIONALE
Provocare clinică? unu
Pacientul R., 25 de ani, a fost internat departamentul terapeutic cu plângeri de erupții petechiale și vânătăi pe pielea extremităților inferioare, suprafața anterioară a corpului, apărute spontan sau din cauza celei mai mici traume, și menoragii, sângerări nazale.
Din istoricul medical se știe că în ultima lună au apărut trei sângerări nazale spontane și vânătăi pe piele după mici vânătăi. În urmă cu 3 zile, după extragerea dintelui „de minte”, a început imediat sângerarea, care a fost oprită abia după 12 ore.În legătură cu acest eveniment, pacienta a decis să fie examinată într-un spital.
Din istoria vieții se știe că în copilărie a avut rujeolă, SARS, ereditatea nu este împovărată, obiceiuri proaste neagă.
Obiectiv: starea este relativ satisfăcătoare, pe pielea extremităților inferioare și pe suprafața anterioară a corpului apar erupții petehiale care nu ies peste suprafața pielii, nedureroase la apăsare, și o cantitate mică de vânătăi de formă neregulată.
Nu a fost găsită nicio patologie în alte organe și sisteme.
Date metode de laborator cercetare.
Test clinic de sânge: Hb - 120 g/l, leucocite - 6,5x10 9/l, eritrocite - 4,5x10 11/l, trombocite - 50.000/l. Analiza biochimică sânge fără patologie. VSC este norma. APTT este normal. VK - 4,5 min. 1. Formulați un diagnostic preliminar.
2. Ce indicatori din testele screening ale tulburărilor de hemostază sunt modificați la acest pacient?
3. Realizați un plan de tratament consecvent pentru acest pacient.
4. Determinați tipul de sângerare la acest pacient.
9. TESTE FINALE
Alegeți unul sau mai multe răspunsuri corecte.
1. Selectați semnele caracteristice hemofiliei A:
B. Hematom tip de sângerare.
G. Tipul de sângerare cu pete petehiale. D. Testul manșetei pozitiv.
2. Hemofilia A se caracterizează prin:
A. Prelungirea timpului de sângerare. B. Timp normal de sângerare.
B. Prelungirea timpului de coagulare a sângelui. D. Timp normal de coagulare. D. Număr normal de trombocite.
3. Hemofilia se caracterizează prin toate, cu excepția:
A. Prelungirea APTT.
B. Număr normal de trombocite.
B. Oprirea hemoragiei din cauza transfuziei de plasmă proaspătă congelată.
G. Hematom-tip petechial de sângerare. D. Natura ereditară a bolii.
4. Următoarele semne sunt caracteristice hemofiliei:
A. Reducerea numărului de trombocite. B. Hemartroze cronice.
B. Vasculita-tip de sângerare violet. G. Hematom tip de sângerare.
D. Efectul pozitiv al splenectomiei.
5. Următoarele simptome nu sunt tipice pentru hemofilie:
A. Formarea hematomului în injecție intramusculară medicamente.
B. Sângerări „târzii” la 2-3 ore după ce se opresc.
B. Număr normal de trombocite. D. Mărirea ficatului și a splinei.
D. Reducerea vieții trombocitelor.
6. Selectați semnele caracteristice trombocitopeniei:
A. Tipul de sângerare cu pete petechiale. B. Hematom-tip petechial de sângerare.
B. Timp normal de sângerare. D. Prelungirea timpului de sângerare.
D. Durata normală de viață a trombocitelor.
7. Următoarele semne sunt caracteristice trombocitopatiei:
A. Reducerea numărului de trombocite. B. Număr normal de trombocite.
B. Scăderea timpului de coagulare.
D. Efectul pozitiv al tratamentului cu corticosteroizi. D. Timp normal de sângerare.
8. Selectați cauzele trombocitopeniei:
A. Distrugerea crescută a trombocitelor. B. Consum crescut de trombocite.
B. Icter hemolitic.
G. Leziuni ischemice ale organelor. D. Concentrație mare fibrinogen.
9. Selectați medicamente care nu sunt indicate pentru pacienții cu trombocitopenie:
A. Citostatice. B. Hormoni.
D. Salicilati. D. Vitamine.
10. Selectați teste pentru screeningul diagnosticului de trombocitopenie și trombocitopatii:
A. Determinarea numărului de trombocite. B. Studiul aderenței trombocitelor.
B. Studiul agregării trombocitelor. D. Determinarea timpului de sângerare.
D. Determinarea timpului de coagulare a sângelui.
11. Corelați tipurile de sângerare cu manifestările lor externe prezentate în figuri (vezi insert, p. 5, fig. 8-1):
A. Hematom.
B. Purpuriu vasculitic.
B. Petehial-pătat. G. Hematom petehial.
D. Angiomatos.
12. Selectați simptomele caracteristice bolii Schonlein-Genoch:
A. Erupție cutanată simetrică pe extremități.
B. Hemartroza simetrică a articulațiilor mari.
B. Microhematurie.
D. Lipsa pigmentării după erupție cutanată. D. Pigmentare brună după erupție cutanată.
13. Vasculita hemoragică se caracterizează prin afectarea vaselor de următoarea localizare:
A. Vasele pielii. B. Vasele rinichilor.
B. Vasele hepatice.
G. Vasele splinei. D. Vasele intestinale.
14. Pacienții cu vasculită hemoragică se caracterizează prin:
A. Reducerea numărului de trombocite. B. Mărirea splinei.
B. Mărirea ficatului.
G. Splina dimensiuni normale. D. Ficat de dimensiuni normale.
15. Boala Schonlein-Genoch se poate manifesta:
A. Deformarea grosieră a îmbinărilor. B. Dezvoltarea insuficientei renale cronice.
B. Recurente sângerare uterină. G. Simptome de „abdomen acut”.
D. Splenomegalie.
16. Pentru tratamentul bolii Schönlein-Genoch se folosesc următoarele:
A. Antihistaminice. B. Vitamina K.
B. Preparate de calciu. D. Vitamina C.
D. Niciuna dintre cele de mai sus.
17. Telangiectazia hemoragică ereditară (boala Rendu-Osler) se caracterizează prin următoarele simptome:
A. Reducerea numărului de trombocite. B. Hemartroză simetrică.
B. Sângerări nazale persistente recurente. D. Erupții petehiale pe piele.
D. Apariția angioamelor în copilărie.
18. Boala Von Willebrand se caracterizează prin:
A. Hematom tip de sângerare.
B. Hematom petehial tip de sângerare.
B. Reducerea numărului de trombocite. D. Număr normal de trombocite.
D. Defect în legătura de coagulare a hemostazei.
19. Selectați semne care nu sunt tipice pentru boala Rendu-Osler:
A. Mărirea splinei.
B. Sângerări persistente recurente (nazale, uterine etc.).
B. Efectul pozitiv al splenectomiei.
D. Efectul pozitiv al tratamentului cu corticosteroizi. D. Necesitatea examenului endoscopic diagnostic suplimentar.
20. Pentru oprirea sângerării în boala Randu-Osler sunt indicate următoarele:
A. Vitamina K.
B. Acid aminocaproic.
B. Clorura de calciu.
D. Aplicarea locală a soluției de trombină. D. Administrarea sistemică a heparinei.
10. STANDARDE DE RĂSPUNSURI
10.1. Răspunsuri la sarcinile de testare ale nivelului inițial
10.2. Răspunsuri la sarcini situaționale
Provocare clinică? unu
1. Pacientul are probabil purpură trombocitopenică (fără ereditate agravată, fără manifestări ale bolii în copilărie, fără mărire a ficatului și a splinei, scăderea numărului de trombocite la 50.000).
2. Tipul de sângerare cu pete petehiale.
3. Pacientul va avea timp de sângerare prelungit.
4. Este indicat tratamentul cu corticosteroizi. Daca in 3-4 luni tratamentul cu corticosteroizi este ineficient, este indicata splenectomia, iar daca aceasta din urma este ineficienta este indicata o combinatie de citostatice si corticosteroizi.
10.3. Răspunsuri la sarcinile de testare finale
11. A - 2; B - 1.
Sindromul hemoragic este o afecțiune patologică a corpului unui adult sau al unui copil. Se exprimă prin sângerare crescută. Termenul în sine nu este o definiție a unei anumite boli. Sindromul hemoragic este considerat o manifestare a unui grup de boli care au motive diferite dezvoltare, dar sunt conectate prin simptome similare și mecanismul dezvoltării.
Un alt nume pentru afecțiune este diateza hemoragică. În plus, sunt luate în considerare caracteristicile patologiei, principalele forme, factorii și cauzele dezvoltării, principiile tratamentului.
Starea de sângerare crescută la copii și adulți se dezvoltă din următoarele motive:
- patologia coagulării sângelui (coagulopatie congenitală sau dobândită);
- o scădere a indicatorilor cantitativi sau o încălcare a structurii celulelor plachetare (trombastenie, trombocitopenie);
- deteriorarea pereților vaselor de sânge (vasculită hemoragică);
- o combinație a mai multor dintre factorii de mai sus (boala von Willebrand).
Important! Sindromul hemoragic include mai multe boli, prin urmare are diverse motive apariția.
Ereditate
![]() Există, de asemenea, factori ereditari în dezvoltarea sindromului. Un exemplu de astfel de boală este hemofilia. Aceasta este o coagulopatie de natură congenitală, în care nu există anumiți factori de coagulare a sângelui. Boala se manifestă la 1 nou-născut din 50 de mii de cazuri.
Există, de asemenea, factori ereditari în dezvoltarea sindromului. Un exemplu de astfel de boală este hemofilia. Aceasta este o coagulopatie de natură congenitală, în care nu există anumiți factori de coagulare a sângelui. Boala se manifestă la 1 nou-născut din 50 de mii de cazuri.
Un alt exemplu al rolului eredității este boala von Willebrand. Afecțiunea este asociată cu un defect genetic al factorului de coagulare von Willebrand, care este implicat în aderența trombocitelor. Telangiectazia - poate fi congenitală sau dobândită. Ereditatea iese pe primul loc dacă această afecțiune este un simptom al bolii Rendu-Osler, al sindromului Louis-Bar, al angiomatozei.
Boli dobândite
Lista stărilor patologice care se dezvoltă de-a lungul vieții nu au nicio legătură cu anomalii congenitale:
- una dintre formele de trombocitopatie;
- DIC;
- una dintre formele vasculitei hemoragice.
Provocatorii de boli pot fi infecții virale, utilizare pe termen lung medicamente (sulfonamide, butadionă, chinină), leziuni grave, condiții de șoc, transfuzie de sânge.
Simptomele sindromului hemoragic
Tabloul clinic depinde de boala care a cauzat dezvoltarea tulburărilor.
Hemofilie
Sângerarea crescută se observă chiar și la nou-născuți. Se exprimă prin hemoragii și vânătăi. Ele apar la cea mai mică vânătăi, răni, tăieturi. Sângerarea se dezvoltă chiar și pe fondul unei modificări a dinților de lapte.
Cu leziuni mecanice și orice intervenții apar hemoragii în articulații, împotriva cărora mai târziu procese inflamatorii, mobilitate limitată. Sângerare internă și periculoasă, apariția sângelui în urină.
Trombocitopenie autoimună (boala Werlhof)
 Simptomele bolii apar dacă nivelul trombocitelor din sânge scade la 50 * 109 / l. Cursul poate avea o formă acută sau cronică încet progresivă.
Simptomele bolii apar dacă nivelul trombocitelor din sânge scade la 50 * 109 / l. Cursul poate avea o formă acută sau cronică încet progresivă.
Pe piele apar hematoame petehiale, se dezvoltă sângerări în diferite grade din membranele mucoase. La examinare, splina este mărită.
Boala Henoch-Schonlein (vasculită hemoragică)
Simptomele sindromului hemoragic pe fondul vasculitei depind de forma acestuia.
- Cutanat - hemoragii cu puncte mici pe pielea feselor, a extremităților inferioare, care se caracterizează prin simetrie. Peteșiile se pot inflama, lăsând zone cu pigmentare crescută, nu mâncărime și nu provoacă disconfort. După 5 zile, manifestările sunt reduse.
- Articular - durere, roșeață, umflare pe suprafețele articulațiilor. Procesele sunt reversibile.
- Abdominal – caracterizat hemoragii interneși sângerare. Sindromul hemoragic se manifesta prin faptul ca pacientii se plang dureri severeîn abdomen, febră, vărsături, sânge în scaun.
- Renală – se dezvoltă în funcție de tipul de nefrită sau insuficiență cronică rinichi.
- Curge rapid - hemoragie în țesutul cerebral.
boala von Willebrand
Sindromul hemoragic se manifestă prin sângerare prelungită. Această formă a bolii este cea mai periculoasă pentru copii și adulți, deoarece toate cele trei verigi în procesul de oprire a sângerării (hemostaza) sunt încălcate în mecanismul dezvoltării sale.
Formele ușoare ale sindromului se manifestă prin sângerări nazale minore, hemoragii cutanate. Severă - sângerare masivă prelungită de diferite localizări ( organe interne, articulații, țesuturi moi).
Notă! La examinare, specialiștii găsesc adesea la pacienți vase dilatate și sinuoase, care sunt localizate sub formă de plexuri.
DIC
Alte denumiri pentru sindrom sunt sindromul trombohemoragic, coagulopatia de consum. Ar putea avea dezvoltare acută sau scurge fără vizibil semne clinice. Încălcarea apare cu patologii obstetricale, stări de șoc, leziuni severe, sepsis de origine bacteriană.
Apare sindromul hemoragic următoarele simptome:
- Etapa I - există semne de tromboză masivă, o scădere a volumului de sânge circulant în corpul pacientului, o încălcare a proceselor metabolice.
- Etapa II - afectarea multor organe, blocarea alimentării cu sânge a ficatului, pancreasului, plămânilor, creierului, glandelor suprarenale, splinei. Erupție cutanată petechială pe piele combinate cu hemoragii hemoragice de diverse localizari.
- Stadiul III - pacientul dezvoltă insuficiență respiratorie, insuficiență cardiacă, hepatică, renală, apare pareza intestinală. Sângerare masivă și hemoragie.
- Etapa IV - cu un rezultat favorabil al patologiei, toate funcțiile corpului sunt restabilite, în caz contrar pacientul moare.
Important! Unul dintre motivele dezvoltării DIC este febra hemoragică (are natură infecțioasă).
 În primul rând, specialiștii colectează o anamneză a vieții și bolii pacientului. Medicii precizează prezența rudelor bolnave, perioada în care sindromul hemoragic și-a început manifestările, prezența transfuziilor de sânge în trecut, infecții și leziuni. Pacientul află ce factori, în opinia sa, au determinat dezvoltarea patologiei.
În primul rând, specialiștii colectează o anamneză a vieții și bolii pacientului. Medicii precizează prezența rudelor bolnave, perioada în care sindromul hemoragic și-a început manifestările, prezența transfuziilor de sânge în trecut, infecții și leziuni. Pacientul află ce factori, în opinia sa, au determinat dezvoltarea patologiei.
Medicii evaluează aspect pacient, plângerile lui. Se confirmă dezvoltarea sindromului hemoragic diagnostic de laborator. ÎN analiza generala sângele fixează nivelul tuturor elemente de formă, hemoglobina. O atenție deosebită trebuie acordată indicatorilor cantitativi ai trombocitelor.
Se efectuează o coagulogramă pentru a clarifica momentul coagulării sângelui, durata sângerării. Evaluați fibrinogenul și protrombina, sensibilitatea la heparină.
Important! Metode suplimentare sondajele sunt analiza clinica urină, biopsie măduvă osoasă, străpungere măduva spinării, teste genetice.
Sindromul hemoragic la nou-născuți
O caracteristică a nou-născuților este tendința de a crește sângerarea. Acest lucru este asociat cu particularitățile sistemului de coagulare a sângelui. Specialiștii disting două grupuri de copii cu astfel de tulburări:
- un copil „sănătos” a cărui creștere a sângerării este o afecțiune fiziologică;
- Un copil „bolnav” al cărui sindrom hemoragic are cauze patologice.
Clasificarea sindromului hemoragic la nou-născuți:
- Coagulopatie - hemofilie, boala hemoragica, DIC-sindrom, patologii pe fondul bolilor tract intestinal, ficat.
- purpură trombocitopenică.
- Trombocitopatie (congenitală, ereditară).
- Tulburări vasculare - simptomele sindromului hemoragic sunt combinate cu indicatori normali sisteme de coagulare.
Boala hemoragică a nou-născutului
 Cauza afecțiunii este lipsa factorilor dependenți de vitamina K, adică tulburările apar pe fondul deficienței de vitamina K în corpul pacientului. Alăptarea unui copil în primele ore după naștere este o prevenire eficientă a dezvoltării sindromului.
Cauza afecțiunii este lipsa factorilor dependenți de vitamina K, adică tulburările apar pe fondul deficienței de vitamina K în corpul pacientului. Alăptarea unui copil în primele ore după naștere este o prevenire eficientă a dezvoltării sindromului.
În cazul stadiului inițial, simptomele bolii apar în prima zi după naștere. De regulă, dezvoltarea este asociată cu acceptarea maternă medicamentele(barbiturice, antiinflamatoare nesteroidiene, warfarină, unii agenți antibacterieni).
Sindromul hemoragic în forma sa clasică apare în prima săptămână de viață a unui copil. Există hemoragii pe piele, sângerări de diferite locații, o scădere bruscă tensiune arteriala, dificultăți de respirație, scaun negru (melena).
Important! Formele tardive ale stării patologice la copii apar în primele 1,5 luni de viață. Simptomele sunt similare cu manifestarea clasică.
Coagulopatie ereditară
Dezvoltarea sindromului hemoragic de tipul hemofiliei B și a bolii von Willebrand practic nu are loc la nou-născuții. Simptomele vii pot apărea numai cu hemofilia A (și apoi doar la jumătate din băieții bolnavi).
Tratamentul sindromului hemoragic
Dacă se dezvoltă un sindrom hemoragic, pacientului trebuie să i se acorde primul ajutor. Tratamentul suplimentar se efectuează într-un spital sau într-o unitate de terapie intensivă (în funcție de severitate).
Primul ajutor
Constă din următoarele activități:
- Opriți sângerarea - utilizați bandaj de presiune(din venă), turunda (din nas), pungă cu gheață (internă), garou (din arteră).
- Medicamente hemostatice (Dicinon, Etamzilat, Vikasol) - doza este determinată de un specialist.
- Introducerea acidului aminocaproic într-o venă prin jet sau picurare.
- Perfuzii de înlocuitori ai sângelui (soluții saline, dextrani, preparate plasmatice).
- Controlul tensiunii arteriale, al temperaturii corpului, al pulsului, al respirației.
- Spitalizarea la hemoragie internă, dintr-o venă, arteră, orice alta care nu poate fi oprită în afara spitalului.
Tratament
Tratamentul depinde de starea patologică și de motivele care au determinat dezvoltarea acesteia.
Cu purpura trombocitopenică, se prescrie o dietă, repaus strict la pat. Baza tratamentului preparate hormonaleîn tablete în doză de 2 mg/kg pe zi. Cursul este de 14 zile. Pentru a opri sângerarea, se prescriu acid aminocaproic, Dicinon, Etamzilat. La stare gravă- transfuzie de trombocite. Dacă este necesar, îndepărtați splina.
Tratamentul bolii Shenlein-Genoch se bazează pe dieta terapie, prescrierea de medicamente. Dintre medicamente, se folosesc antioxidanți, heparină, medicamente antialergice, hormoni. Dacă este necesar - citostatice și antibiotice (doza de fonduri este selectată individual).
Tratamentul hemofiliei se bazează pe introducerea de crioprecipitat, concentrat de plasmă nativă, plasmă proaspătă congelată (doza de substanțe de înlocuire este determinată de medic).
Prevenirea sindromului hemoragic
Se compune din următoarele:
- dieteterapie cu o cantitate mare vitamina K și proteine;
- excluderea daunelor, vătămării;
- consultație cu un genetician pentru cuplurile care intenționează să conceapă un copil;
- examinarea periodică a pacienților și examinarea clinică oameni sanatosi;
- tipuri permise de terapie cu exerciții fizice;
- administrarea profilactică a factorilor sanguini.
Boli și sindroame hemoragice - stări patologice, caracterizată prin sângerare crescută ca urmare a insuficienței unuia sau mai multor elemente de hemostază. În acest articol, ne vom uita la principalele semne și simptome ale bolilor hemoragice la oameni.
Boli și sindroame hemoragice
Semne de boli hemoragice
Sângerarea în telangiectazii hemoragice ereditare se datorează dezvoltării insuficiente a cadrului subendotelial al vaselor mici și inferiorității endoteliului în anumite zone ale patului vascular. într-o creșă sau adolescent se formează angioame mici cu pereți subțiri, ușor de accidentat; în unele cazuri, șunturile arteriovenoase se formează și în plămâni și în alte organe. Inferioritatea țesuturilor mezenchimale se poate manifesta și prin extensibilitate crescută a pielii („piele de cauciuc”), slăbiciune a aparatului ligamentar (luxații obișnuite, prolaps al cuspidelor valvelor cardiace). Rareori, această boală duce la deces din anemie acută post-hemoragică, când, de exemplu, bandajarea arterelor carotide nu se poate opri sângerare din nas. Poate o combinație cu boala von Willebrand.
Simptomele bolilor hemoragice
Boala se manifesta prin sangerari recurente din telangiectazii, localizate cel mai adesea in cavitatea nazala. Mai rar, telangiectaziile sângerează pe marginea buzelor, a mucoaselor cavității bucale, a faringelui și a stomacului. Numărul de telangiectazii (și, în consecință, sângerare de la acestea) crește în timpul pubertății și la vârsta de 20-30 de ani. Odată cu formarea șunturilor arteriovenoase, se pot dezvolta dificultăți de respirație, cianoză și eritrocitoză hipoxică. Poate o combinație cu prolapsul cuspidelor valvelor cardiace (suflu, aritmie), hipermobilitatea articulațiilor, luxații și alte tulburări cauzate de inferioritatea țesuturilor mezenchimale, precum și cu o deficiență a factorului von Willebrand. Se poate dezvolta ciroza hepatică.
Forme de boli și sindroame hemoragice
Alocați forme ereditare și dobândite de boli și sindroame hemoragice.
Formele ereditare sunt asociate cu determinate genetic modificări patologice perete vascular, anomalii ale megacariocitelor, trombocitelor, proteinelor adezive plasmatice și factorilor plasmatici ai sistemului de coagulare a sângelui.
Formele dobândite în majoritatea cazurilor se datorează:
- afectarea vaselor de sânge de etiologie imună, imunocomplex, toxicoinfectios și dismetabolic;
- afectarea megacariocitelor, trombocitelor de diferite etiologii;
- patologia proteinelor adezive ale plasmei sanguine și a factorilor sistemului de coagulare a sângelui;
- tulburări multifactoriale ale sistemului de coagulare a sângelui ( sindroame acute GHEAŢĂ).
Clasificarea bolilor hemoragice
În funcție de patogeneză, se disting următoarele grupuri de boli și sindroame hemoragice:
Condiţionat leziune primară perete vascular cu posibilă dezvoltare secundară a coagulării și a tulburărilor plachetare. Acest grup include telangiectazia hemoragică ereditară Randu-Osler, sindromul Ehlers-Danlos, sindromul Marfan, hemangioamele gigantice în sindromul Kazabakh-Merritt, vasculita hemoragică Schoenlein-Genoch, eritemul, febra hemoragică C și hipovitaminoza B, etc.
Cauzată de leziunea primară a germenului megacariocito-trombocitar.
trombocitopenie: redistribuirea trombocitelor și depunerea lor în splină; distrugere crescută (de exemplu, în LES; acest grup include și purpura trombocitopenică idiopatică); consum crescut de trombocite și formare de trombi (DIC, purpură trombocitopenică trombotică); utilizarea unor medicamente.
Trombocitopatii: afecțiuni caracterizate prin trombocite anormale și/sau o încălcare a funcțiilor acestora. Cele mai frecvente dintre ele sunt trombastenia Glanzmann și boala von Willebrand.
Cauzată de tulburări de coagulare a sângelui (coagulopatie):
- coagulopatie ereditară: hemofilie A, hemofilie B, boala von Willebrand, deficit de factori de coagulare a sângelui;
- coagulopatii dobândite: coagulopatii dependente de vitamina K (apar cu insuficiență hepatică, malabsorbție a vitaminei K, deficiență nutrițională a vitaminei K, administrarea de medicamente precum cumarina), DIC, patologie hepatică (conduce la o deficiență a multor factori de coagulare), inhibitori patologici de coagulare anticoagulant ("lupus"; inhibitori specifici de coagulare - AT, specifici proteinelor individuale de coagulare);
- încălcări ale stabilizării fibrinei, creșterea fibrinolizei, inclusiv tratamentul cu anticoagulante directe și indirecte, fibrinolitice (streptokinază, urokinază, alteplază etc.);
- alte tulburări de coagulare dobândite: deficienţa factorilor de coagulare poate apărea când boli somatice(de exemplu, în amiloidoză - deficit de factor X).
Cauzat de tulburări complexe ale diferitelor părți ale sistemului de coagulare a sângelui (sindroame acute DIC).
Ca grup special, se disting diferite forme ale așa-numitei sângerări artificiale cauzate de pacienții înșiși (de exemplu, cu probleme mentale) prin traumatizarea mecanică a țesuturilor (smulgerea sau suptul vânătăilor, traumatisme ale mucoaselor etc.), luarea de medicamente hemoragice (cel mai adesea anticoagulante indirecte: cumarine, fenilina etc.), autotortura sau sadismul.
Sângerări în bolile hemoragice
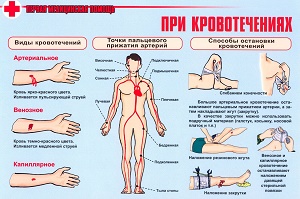
Există următoarele tipuri de sângerare:
Capilară sau microcirculatoare (petehial-echimoze) - tipul de sângerare se caracterizează prin erupții petehiale, vânătăi și echimoze pe piele și mucoase. Adesea combinat cu sângerare crescută a membranelor mucoase (sângerări nazale, menoragie). Posibilă dezvoltare a hemoragiilor severe la nivelul creierului. Acest tip de sângerare este caracteristic trombocitopeniei și trombocitopatiilor, bolii von Willebrand, deficienței factorilor complexi de protrombină (VII, X, V și II), unele variante de hipo- și disfibrinogenemie, supradozaj moderat de anticoagulante. În cazul trombocitopatiilor ereditare, se observă de obicei un tip de sângerare cu vânătăi, o erupție petechială nu este tipică.
Hemoragiile de tip hematom se caracterizează prin hemoragii dureroase, intense la nivelul țesutului subcutanat, mușchi, articulații mari, în peritoneu și spațiu retroperitoneal. Hematoamele pot duce la compresia nervilor, distrugerea cartilajului și țesut osos, disfuncție a sistemului musculo-scheletic. Uneori se dezvoltă sângerări renale și gastrointestinale. Sângerarea prelungită este caracteristică tăieturilor, rănilor, după extracția dinților și interventii chirurgicale conducând adesea la anemie. Acest tip de sângerare se observă în unele tulburări ereditare de coagulare a sângelui (hemofilie A și B, deficiență severă a factorului VII), coagulopatie dobândită, însoțită de apariția inhibitorilor factorilor VIII, IX, VIII + V în sânge și cu o supradoză de anticoagulante, precum și în trombocitopatia ereditară cu absența factorului de placa a 3-a.
Hemoragiile de tip capilar-hematom mixt se caracterizează prin erupții cutanate petehiale, combinate cu hemoragii dense extinse și hematoame. Se observă în forme ereditare (deficiență severă a factorilor VII și XIII, formă severă a bolii von Willebrand) și dobândite (sindroame acute DIC, supradozaj semnificativ de anticoagulante indirecte) încălcări.
Tipul de sângerare vasculitic-violet se caracterizează prin erupții cutanate hemoragice sau eritematoase (pe bază inflamatorie), este posibilă dezvoltarea nefritei și sângerării intestinale; observată în vasculitele infecțioase și imune.
Hemoragia de tip angiomatos se caracterizează prin sângerare persistentă, strict localizată, asociată cu patologia vasculară locală. Se observă cu telangiectazi, angioame, șunturi arteriovenoase.
Diagnosticul bolilor hemoragice
Diagnosticul general al bolilor și sindroamelor hemoragice se bazează pe următoarele principii de bază:
Determinarea momentului de debut, prescripție, durata bolii și caracteristicile cursului acesteia: debut la o vârstă fragedă sau la adulți, dezvoltarea acută sau treptată a sindromului hemoragic, curs recent sau pe termen lung (cronic, recurent).
Identificarea, dacă este posibil, a genezei ereditare a sângerării (cu specificarea tipului de moștenire) sau a naturii dobândite a bolii. Clarificarea posibilei legături dintre dezvoltarea sindromului hemoragic și anterioare procese patologice, influențe (inclusiv cele terapeutice - consumul de medicamente, vaccinare etc.) și boli de fond (boli hepatice, leucemie, procese infecțio-septice, leziuni, șoc).
Determinarea localizării predominante, a severității și a tipului de sângerare. De exemplu, în boala Rendu-Osler, predomină sângerările nazale persistente (adesea aceasta este singura manifestare clinică); cu patologia trombocitară - tendință crescută la echimoze, sângerări uterine și nazale, cu hemofilie - hematoame profunde și hemoragii la nivelul articulațiilor.
La diagnostic diferentiat trebuie luate în considerare diferențele semnificative în prevalența bolilor hemoragice individuale: unele dintre ele sunt extrem de rare, în timp ce altele reprezintă marea majoritate a celor întâlnite în practica clinica cazuri de sângerare.
Telangiectazii hemoragice ereditare
Telangiectazia hemoragică ereditară (angiomatoza hemoragică, boala Rendu-Osler) se moștenește în mod autosomal dominant.
Laborator și metode instrumentale . Sângerările frecvente și abundente pot duce la dezvoltarea anemiei posthemoragice. Odată cu formarea șunturilor arteriovenoase, se poate dezvolta eritrocitoză, o creștere a concentrației de Hb în sânge; la examinare cu raze X plămânii prezintă umbre unice rotunjite sau de formă neregulată, adesea confundate cu tumori.
Diagnosticul se bazează pe identificarea naturii familiale a bolii (cu toate acestea, sunt posibile și cazuri sporadice) și pe detectarea telangiectaziei tipice și a sângerărilor recurente din acestea.

