23.02.2018
Arnold chiari complicație și cauze. Anomalia Arnold-Chiari: cauze ale malformației, manifestări, diagnostic, tratament, prognostic
Anomalia lui Arnold - Chiari - o tulburare congenitală a structurii unei anumite părți a creierului, numită romboid. Acest lucru duce la o deplasare a structurilor creierului și la perturbarea fluxului de lichid cefalorahidian (LCR). Există mai multe tipuri de dezvoltare a patologiei, care determină severitatea cursului bolii, prognosticul acesteia. Tratat de neurologi și neurochirurgi.
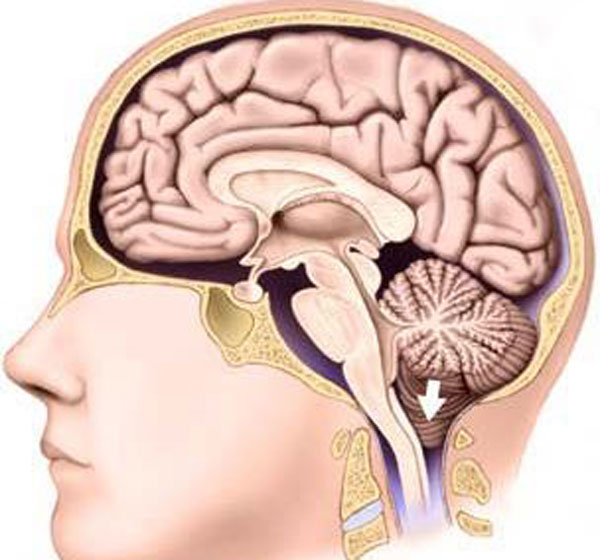
În neurologie, există conceptul de „creier romboid”. Aceasta este medulul oblongata, cerebelul și structura situată între lobii acestuia din urmă, numită „punte”. Toate aceste departamente ocupă o depresiune pereche pe baza osoasă a craniului - fosa craniană posterioară. În centrul acestuia din urmă se află cea mai mare gaură - un occipital mare. Este cea care conectează cavitatea craniană cu canalul spinal. Trecând prin această deschidere, medulla oblongata, însoțită de vase mari și nervi, devine măduva spinării.
Suprafețele medulei oblongate și pons cu fața către oasele occipitale formează o depresiune specifică - o fosă romboidă. Prin el, lichidul cefalorahidian trece de la creier la măduva spinării.
Odată cu anomalia Arnold-Chiari, dimensiunile recipientului osos nu mai corespund volumului structurilor aflate în acesta. Ca urmare, există mai puțin spațiu pentru creierul romboid și trebuie să se deplaseze în foramen magnum până la nivelul vertebrelor cervicale I-II.
O astfel de dislocare duce la o încălcare mai mult sau mai puțin puternică. Trunchiul cerebral și rădăcinile nervilor cranieni sunt compensatorii, pentru a nu fi strânse de oase (ceea ce va duce la moarte), se lungesc. În plus, cerebelul apăsat pe trunchi blochează fluxul liber al lichidului cefalorahidian, în urma căruia se dezvoltă.
În cazurile severe, anomalia Chiari este asociată cu o hernie. măduva spinării, formarea de cavități în medula oblongata și măduva spinării.
Cauze
Etiologia anomaliei nu a fost încă stabilită. Se crede că dezvoltarea sa se datorează defectelor congenitale ale sistemului osos și ale sistemului nervos în același timp. Se crede că în prezența anomaliilor craniene și cerebrale „programate”, pentru ca anomalia Chiari să se dezvolte, trebuie să fie prezenți încă 2 factori:
- deteriorarea unei structuri osoase numită clivus osul sfenoid, care se află în fața foramenului occipital mare (ca urmare, devine mai larg), la naștere;
- un „val” de putere crescută a lichidului cefalorahidian care lovește simultan canalul central al măduvei spinării (o mică gaură în centrul substanței sale cenușii).
Grade
Există 4 tipuri de această anomalie.
eu scriu
Acesta este cel mai frecvent tip de patologie. În acest caz, doar amigdalele (lobuli inferiori) cerebelului pe una sau ambele părți trec prin deschiderea mare occipitală și intră în canalul spinal. Patologia începe să apară abia la vârsta de 30-40 de ani, de unde și numele - tipul de anomalie „adult”.
tip II
În același timp, atât amigdalele cerebelului, cât și structura situată între ele (vermisul cerebelos) trec prin foramen magnum. Medula oblongata și al patrulea ventricul sunt, de asemenea, deplasate. Aceasta determină dezvoltarea hidrocefaliei mai mult sau mai puțin pronunțate.
Cu acest tip, există o hernie în regiunea lombară (o secțiune a măduvei spinării iese între vertebre, acoperită cu membrane), există defecte de dezvoltare OS occipitalși vertebrele cervicale.
tipul III
Toate structurile localizate în fosa craniană posterioară sunt deplasate în canalul rahidian. Există o hernie cerebrală (cerebelul și, uneori, lobul occipital al creierului din membrane iese) în regiunea occipitală sau cervicală.
tip IV
Aceasta este o scădere a dimensiunii cerebelului fără luxația acestuia.
Principalele simptome ale anomaliei sunt următoarele:
- Dureri în partea din spate a capului și a gâtului, care va crește odată cu tusea, încordarea și strănutul. Este cel mai puternic dimineața.
- Strănutul, râsul, tusea și încordarea pot provoca, de asemenea, o dezvoltare ascuțită a amorțelii pe jumătate din corp, „pielea de găină” în mâini.
- Scăderea temperaturii și sensibilitatea la durere în mâini.
- Mușchii brațelor sunt slabi.
- Convulsii.
- Cu siringomielia existentă, sensibilitatea scade segmentat.
Diagnosticul se face numai pe baza celor efectuate, care dezvăluie:
- localizarea amigdalelor cerebeloase la 3 sau mai mulți milimetri sub nivelul foramenului magnum;
- reducerea volumului amigdalelor cerebelului;
- compresia părții inferioare a medulei oblongate și a părții superioare a măduvei spinării de către amigdalele cerebelului;
- hidrocefalie;
- prezența cavităților în medula oblongata și/sau măduva spinării.
Dacă se suspectează siringomielie și există contraindicații pentru un RMN, se poate face un alt studiu, mielografia. Aceasta este o metodă invazivă cu raze X, când un agent de contrast cu raze X este injectat în lichidul cefalorahidian, abia atunci se fac o serie de fotografii.
Tratament
Abordarea tratamentului patologiei este individuală:
- dacă o persoană nu prezintă simptome (anomalia a fost o constatare la un RMN), persoana nu primește niciun tratament medical sau chirurgical, face doar un RMN în fiecare an și se consultă cu un neurochirurg;
- dacă există doar durere în spatele capului și a gâtului, se prescriu analgezice ("Meloxicam", "Nimesulide", "Nurofen") și relaxante musculare ("");
- dacă o persoană are o încălcare a înghițirii sau a respirației, se efectuează o operație urgentă, al cărei scop este eliminarea compresiei medulei oblongate și a măduvei spinării;
- dacă există tulburări de coordonare, leșin, pierderea auzului și pierderea vederii, tratamentul este inițial conservator (se folosesc medicamente adecvate), dacă nu există efect, se efectuează o operație.
Cel mai adesea, în timpul operației, se realizează o creștere a volumului fosei craniene posterioare prin îndepărtarea unei secțiuni a osului occipital. Intervenția chirurgicală durează aproximativ 2 ore, rămânerea în spital după aceea este indicată 5-7 zile.
Prognoza
Cu tratamentul în timp util al patologiei de tip I și II, se pot obține rezultate bune până la dispariția completă a deficitului neurologic. Prognosticul anomaliilor de tip III este grav pe viață, chiar și cu o operație de urgență.
18 martie la 13:37 11644 0
Termenul de „malformație Chiari” este preferat față de tradiționala „malformație Arnold-Chiari” datorită contribuției semnificativ mai mari aduse de Chiari.
Malformația Chiari constă din 4 tipuri de anomalii ale creierului posterior, probabil fără legătură. Cele mai multe cazuri sunt în tipurile 1 și 2 (vezi tabelele 6-5). Opțiunile rămase reprezintă un număr extrem de limitat de cazuri.
Tab. 6-5. Caracteristici comparative ale primului și al doilea tip de malformație Chiari (cu modificări)
Malformație Chiari tip 1
așa-zisul. ectopie cerebeloasă primară. Acest anomalie rară, în care există doar deplasarea caudală a cerebelului cu hernie a amigdalelor sub BSO (pentru criterii, vezi RMN mai jos) și „alungirea amigdalelor de tip gag”. Spre deosebire de tipul 2, medula oblongata nu este deplasata caudal (unii autori nu sunt de acord), trunchiul cerebral nu este implicat, nervii cranieni inferiori nu sunt alungiti, iar nervii cervicali superiori nu au directie spre cap.
Poate exista fibroză a membranelor pia și arahnoide din jurul trunchiului cerebral și amigdalelor. Pot exista hidromielie și siringomielie SM. Unele cazuri pot fi dobândite, după șunturi lomboperitoneale sau LP multiple (traumatice).
Epidemiologie
Vârsta medie de manifestare clinică este de 41 de ani (interval: 12-73 de ani). Există o oarecare predominanță a & (&:%=1,3:1). Durata medie simptomatologia asociată definitiv cu malformația Chiari este de 3,1 ani (interval: 1 lună - 20 de ani). Dacă sunt incluse reclamații nespecifice, cum ar fi G/B, termenul ar fi de 7,3 ani. Odată cu apariția erei RMN, această întârziere în diagnosticare poate deveni mai mică.
Manifestari clinice
Pacienții cu o malformație Chiari pot prezenta unul sau toate următoarele simptome:
1. compresia trunchiului cerebral la nivelul BZO
2. GCF
3. siringomielie
4. Separarea regiunii intracraniene de regiunea coloanei vertebrale cu cresteri periodice ale ICP
Reclamații
Cea mai frecventă plângere este durerea (69%), în special G/B, care este de obicei resimțită în regiunea occipitală (Tabelele 6-6).
Tab. 6-6. Plângeri cu malformație Chiari tip 1 (71 de pacienți) 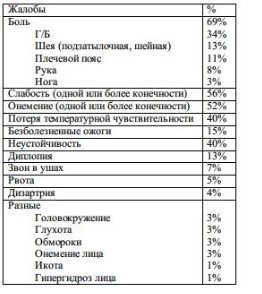
G/B este adesea indus de extensia gâtului și manevra Valsalva. Slăbiciunea este, de asemenea, destul de pronunțată, mai ales cu o strângere de mână unilaterală. Se poate observa simptomul lui Lhermitte. Implicare extremitati mai joase se prezintă de obicei cu spasticitate bilaterală.
Simptome
Vezi tabelul. 6-7. Trei grupuri principale de simptome:
1. Sindrom de compresie BSO (22%): ataxie, tulburări cortico-spinale și senzoriale, simptome cerebeloase, paralizie de insuficiență craniană inferioară. 37% dintre pacienți au H/B severă
2. sindrom leziune centrală SM (65%): tulburări senzoriale disociate (pierderea durerii și a temperaturii cu sensibilitate superficială și profundă păstrată), uneori slăbiciune segmentară și semne de afectare a căilor lungi (sindrom siringomielic). În 11% din cazuri se observă paralizia nervilor cranieni inferiori.
3. sindrom cerebelos (11%): ataxie a trunchiului și a extremităților, nistagmus, disartrie
Tab. 6-7. Simptome ale malformației Chiari tip 1 (121 de pacienți) 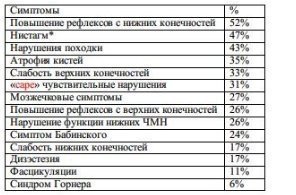
* in cazul clasic: nistagmus descendent cu miscari verticale sau nistagmus rotational cu miscari orizontale; include și oscilopsia
Nistagmusul descendent este considerat caracteristic acestei boli. În 10% din cazuri, starea neurologică a pacienților este neschimbată cu singura plângere de G/B occipital. În unele cazuri, spasticitatea poate fi principala plângere.
curgere naturală
Cursul natural nu este cunoscut cu exactitate (sunt doar 2 publicații pe această temă). Pacientul poate rămâne stabil câțiva ani, cu o deteriorare ocazională. În unele cazuri este posibilă ameliorarea spontană (discutată).
Diagnosticare
Prezentare generală a craniogramelor
În analiza a 70 de craniograme de revizuire s-au constatat modificări doar în 36% din cazuri (în 26% din cazuri a existat o amprentă bazilară, în 7% - platibazie; fiecare pacient avea boala Paget și un clivus concav). Din 60 de spondilograme modificări patologice au fost în 35% din cazuri (inclusiv asimilarea atlasului, extinderea canalului, fuziunea vertebrelor cervicale, ageneza arcului posterior al atlasului).
Metoda de diagnostic la alegere. RMN poate detecta cu ușurință majoritatea anomaliilor de mai sus, inclusiv hernia amigdalelor, precum și hidrosiringomielia, care se observă în 20-30% din cazuri. De asemenea, este vizibilă compresia ventrală a trunchiului cerebral atunci când este prezentă.
Poziția amigdalelor cerebelului în 200 oameni normaliși 25 de pacienți cu malformație Chiari tip 1 sunt prezentate în tabel. 6-8. Tab. 6-9 arată modificările care apar dacă limita poziției inferioare a amigdalelor este considerată a fi 2, și nu 3 mm.

Spre deosebire de informațiile de mai sus, 5 mm este adesea indicată ca limită inferioară a poziției amigdalelor, potrivită pentru recunoașterea prezenței unei malformații Chiari de primul tip.
Mielografie
Fals rezultate negative observată doar în 6% din cazuri. HF ar trebui să urce la BZO.
Datorită artefactelor osoase, CT are limitări în vizualizarea zonei BZO. Fiabilitatea studiului crește atunci când este combinat cu CV administrat intratecal (mielografie). Constatări: omiterea amigdalelor și/sau mărirea ventriculilor.
Tratament
Indicații pentru tratamentul chirurgical
Având în vedere faptul că cele mai bune rezultate se observă atunci când pacienții sunt operați în primii 2 ani de la apariția simptomelor, se recomandă tratament chirurgical precoce pentru pacienții simptomatici. Pacienții asimptomatici pot fi observați și operați ulterior atunci când dezvoltă simptome. Pot fi observați și pacienții care au avut simptome care nu s-au schimbat de câțiva ani. Tratamentul chirurgical pentru acestea este indicat în caz de deteriorare.
Tehnici chirurgicale
Cel mai frecvent tip de intervenție chirurgicală este decompresia PCF (craniectomia suboccipitală) cu sau fără alte intervenții (de obicei combinată cu repararea durei dure și laminectomia cervicală C1 până la C2 sau C3).
Pacientul este așezat pe burtă cu role dedesubt cufăr. Capul este fixat cu un suport pentru cap Mayfield. Gâtul este flectat pentru a mări spațiul dintre occiput și crusul posterior al C1. Umerii sunt trași în jos cu bandă adezivă. O coapsă trebuie așezată pe o rolă (sac de nisip) în cazul în care trebuie să luați un fragment din fascia lata pentru plastie. Incizia se face de-a lungul liniei mediane de la inion la ~ a apofizei spinoase a C5. Osul este rezecat deasupra BZO cu aproximativ 3 cm în sus și cu o lățime de aproximativ 3 cm. DM se deschide cu o incizie în formă de Y și se excizează lamboul superior. ATENŢIE:în malformația Chiari, sinusurile transversale sunt de obicei foarte scăzute (prin urmare, rezecția osului occipital trebuie să fie mică, accentul principal trebuie pus pe laminectomia cervicală și rezecția marginilor BSO pentru a elimina compresia amigdalelor; compresie; a amigdalelor nu apare în PCF). În plus, rezecția excesivă a osului occipital poate duce la bombarea emisferelor cerebeloase în defectul rezultat, care este plin de probleme suplimentare.
Efectuați o grefă durală cu un periost sau un fragment din fascia lata a coapsei pentru a oferi suficient spațiu pentru amigdale și medular oblongata. Unii chirurgi completează operația și cu tamponarea apexului ventriculului IV cu mușchi sau teflon, drenaj al chistului siringomielitic, dacă este prezent (fenestrare, de obicei prin locul de intrare a rădăcinii dorsale, cu sau fără plasarea unui stent sau șunt), Șuntarea ventriculului IV, ventriculostomie terminală, deschiderea orificiului Magendie, dacă este închis).
Unii autori sfătuiesc cu tărie să nu încerce să îndepărteze adeziunile dintre amigdale pentru a evita deteriorarea accidentală a structurilor vitale, inclusiv a PICA. Alții recomandă separarea atentă a amigdalelor și chiar tratamentul lor cu coagulare bipolară pentru a le reduce volumul.
Unii autori recomandă rezecția transorală a clivusului și a procesului odontoid în prezența compresiei ventrale a trunchiului cerebral, tk. ei cred că acești pacienți se pot agrava numai după decomprimarea PCF. Datorită faptului că modificările rezultate s-au dovedit a fi reversibile, pare mai adecvată efectuarea acestei intervenții în cazul deteriorării sau progresiei amprentei bazilare pe RMN după decompresia PCF.
descoperiri operaționale
Vezi tabelul. 6-10. Hernia amigdalelor este prezentă în toate cazurile (prin definiție); cel mai adesea sunt situate la nivelul C1 (62%). În 41% din cazuri, există aderențe între dura mater, membrana arahnoidiană și amigdale cu blocarea orificiilor Luschka și Magendie. În 40% din cazuri, separarea amigdalelor are loc cu ușurință.
Tab. 6-10. Constatări operatorii în malformația Chiari tip 1 (71 de pacienți) 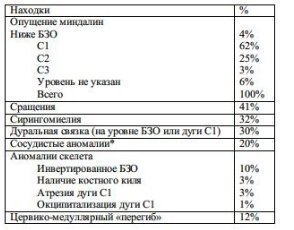
* anomalii vasculare: PICA a fost dilatată sau a avut o localizare neobișnuită la 8 pacienți (PICA coboară adesea până la marginea inferioară a amigdalelor); lacune venoase durale mari
Complicații chirurgicale
După craniectomie suboccipitală plus laminectomie C1-3 la 71 de pacienți (cu repararea duramei la 69 dintre ei), un pacient a murit la 36 de ore după intervenție chirurgicală din cauza apneei în somn. Cea mai frecventă complicație postoperatorie a fost depresia respiratorie (la 10 pacienți), de obicei timp de 5 zile, mai des noaptea. În acest sens, se recomandă monitorizarea atentă a respirației. Alte riscuri ale operației: licoare, hernie ale emisferelor cerebeloase, leziuni vasculare(ZNMA etc.).
Rezultatele operațiunii
Pacienții cu plângeri de durere răspund de obicei bine la intervenții chirurgicale. Slăbiciunea preoperatorie este de obicei mai dificil de tratat, mai ales dacă atrofia musculară este deja prezentă. Senzația se poate îmbunătăți dacă coloanele posterioare nu sunt afectate, iar deficitul se datorează doar implicării tractului spinotalamic.
Roton consideră că principalul efect al operației este oprirea progresiei simptomelor.
Cele mai favorabile rezultate au fost observate la pacienții cu sindrom cerebelos (ameliorare în 87% fără agravare ulterioară). Factori care s-au corelat cu rezultate slabe: prezența atrofiei, ataxiei, scoliozei, durata simptomelor de peste 2 ani.
Tab. 6-11. Rezultate pe termen lung (69 de pacienți, urmărire medie 4 ani) 
* acesti pacienti au avut o deteriorare la nivelul preoperator (fara o deteriorare suplimentara) in 2-3 ani de la operatie; recurența a apărut la 30% dintre pacienții cu sindrom de compresie BSO și la 21% dintre pacienții cu sindrom de leziune SC centrală
Malformație Chiari tip 2
De obicei, combinat cu mielomeningocel (MMC) și mai rar cu o scindare închisă a arcurilor vertebrale (spina bifida oculta).
Fiziopatologia
Probabil nu este asociat cu fixarea SM de către MMC concomitent. Mai probabil, disgeneza primară a trunchiului cerebral cu multe alte anomalii de dezvoltare.
Principalele descoperiri
Luxația caudală a joncțiunii cervico-medulare, a pontului, a ventriculului IV și a medulului oblongat. Amigdalele cerebeloase sunt situate la nivelul BZO sau sub acesta. În loc de îndoirea obișnuită a articulației cervico-medulare, există deformarea acesteia sub formă de inflexiune.
Alte posibile descoperiri:
1. cotul coracoid al plăcii cvadrigeminale
2. Absența septului pellucidum cu aderență intertalamică crescută: se crede că absența septului pellucidum se datorează necrozei cu resorbție de la HCP, mai degrabă decât absenței congenitale.
3. Frunze cerebeloase slab mielinizate
4. GCF: disponibil în majoritatea cazurilor
5. heterotopie (luxație patologică)
6. hipoplazie falx
7. microgirie
8. degenerarea nucleilor nervilor cranieni inferiori
9. anomalii osoase:
A. în zona articulației cervico-medulare
B. asimilare atlas
C. platybasia
D. impresie bazilară
E. Deformare Klippel-Feil
10. hidromielie
11. formarea de goluri în craniu
Manifestari clinice
Manifestările sunt asociate cu disfuncția trunchiului cerebral și a nervilor cranieni inferiori. Manifestarea bolii la vârsta adultă este rară. Manifestările la nou-născuți diferă semnificativ de cele la copiii mai mari. Nou-născuții sunt mai susceptibili de a dezvolta o deteriorare neurologică rapidă cu disfuncție semnificativă a trunchiului cerebral în câteva zile. La copiii mai mari, simptomele se dezvoltă mai treptat și rareori sunt la fel de severe.
Găsește:
1. dificultate la înghițire (disfagie neurogenă) (69%). Se manifestă prin hrănire deficitară, cianoză după hrănire, regurgitare nazală, timp prelungit de hrănire, acumulare de salivă. Reflexul faringian este adesea redus. Mai grav la nou-născuți
2. Apnee (58%): asociată cu stimularea respiratorie afectată. Mai frecvent la nou-născuți
3. stridor (56%): mai frecvent la nou-născuți, de obicei mai rău la inspirație (în timpul laringoscopiei este vizibilă paralizia eferentelor și uneori a adductorilor). corzi vocale) din cauza parezei insuficienței X-a craniană. De obicei temporar, dar poate evolua spre stop respirator
4. aspiratie (40%)
5. slăbiciune membrele superioare(27%), care se poate transforma în tetrapareză
6. opistotonus (18%)
7. nistagmus: mai ales în jos
8. plâns slab sau fără
9. slăbiciune a muşchilor faciali
Diagnosticare
Prezentare generală a craniogramelor
Disproporția cefalofacială poate fi observată ca urmare a HCP congenital. Formarea de goluri la nivelul craniului (așa-numita lukenschadel) se observă în 85% din cazuri (defecte rotunjite ale craniului cu margini clare, separate prin dungi osoase ramificate neuniform). Poate exista o proeminență occipitală internă joasă (PCF scurtată), o creștere a BZO (70%) și o alungire a arcurilor cervicale superioare.
CT și/sau RMN
Principalele descoperiri
A. Deformarea în formă de Z a medulei oblongate*
B. dop cerebelos
C. fuziunea cvadrigeminală (codiunea coracoidală în regiunea cvadrigeminală)
D. creșterea masei intermediare (fuziune intertalamică)*
E. alungirea/cervicalizarea medulei oblongate
F. inserţia joasă a tenonului cerebelos
Descoperiri înrudite
A. GCF
B. siringomielie în zona articulației cervico-medulare (frecvența conform datelor înainte de utilizarea RMN este de 48-88%)
C. ventricul IV dezactivat
D. compresie cerebelo-medulară
E. ageneza/disgeneza corpului calos*
*aceste caracteristici sunt cel mai bine evaluate prin RMN
Laringoscopia
Se utilizează la copiii cu stridor pentru a exclude crup și alte infecții ale tractului respirator superior.
Tratament
Instalați un șunt pentru a corecta GCF (dacă este deja prezent un șunt, verificați funcționarea acestuia)
Cu disfagie neurogenă, stridor, crize de apnee, se recomandă decompresia accelerată a PCF (necesară la 18,7% dintre pacienții cu MMC); asigurați-vă întotdeauna că pacientul are un șunt funcțional înainte de a efectua decompresia recomandată!
Decompresie chirurgicală
NB: Rezultatele operative slabe la nou-născuți se explică prin faptul că multe dintre constatările neurologice se pot datora unor tulburări congenitale (necorectabile) pe care decompresia nu le poate îmbunătăți. Dintr-un alt punct de vedere, modificările histologice se datorează compresiei cronice a trunchiului cerebral și ischemiei concomitente, de aceea decompresia accelerată a trunchiului cerebral trebuie efectuată imediat ce apare oricare dintre următoarele simptome critice de avertizare: disfagie neurogenă, stridor, apnee.
Tehnica de operare: decomprimarea amigdalelor cerebeloase, de obicei cu chirurgie plastică durală. Pacientul este așezat pe burtă, gâtul este îndoit. Produce craniectomie suboccipitală și laminectomie cervicală, care ar trebui să ajungă la vârful amigdalelor cerebelului. Un ligament dural îngroșat, care comprimă SM, poate fi găsit de obicei între marginea BZO și arcul C1. DM se deschide cu o incizie în formă de Y. La deschiderea DM peste nivelul BZO la sugari se impune prudență, deoarece. au sinusul occipital bine dezvoltat și pot avea lacuri durale mari. Nu încercați să separați amigdalele de medulara alungită subiacentă. În cazul în care există o cavitate mare de siringomielita, se efectuează un șunt siringo-subarahnoid.
Dacă există respirație stridor sau pareză a mușchilor abductori laringieni înainte de intervenție chirurgicală, se recomandă o traheostomie (de obicei temporară). Este necesară monitorizarea postoperatorie atentă a respirației (controlul obstrucției și reducerea stimulării ventilației). IVL este indicat pentru hipoxie și hipercarbie.
rezultate
În 68% din cazuri, apare rezolvarea completă sau aproape completă a simptomelor. În 12% din cazuri, există un deficit neurologic rezidual ușor sau moderat. În 20% din cazuri nu există nicio îmbunătățire. În general, rezultatele sunt mai rele la nou-născuți decât la copiii mai mari.
Cel mai cauza comuna mortalitatea este stopul respirator (8 din 17 pacienți decedați). Alte cazuri: meningita/ventriculita (6 pacienti), aspiratie (2) si atrezie biliara (1).
În timpul observației postoperatorii cu durata de la 7 luni la 6 ani, mortalitatea pacienților operați a fost de 37,8%.
Cei mai importanți factori de prognostic au fost starea preoperatorie și rata deteriorării neurologice. Mortalitatea în rândul sugarilor cu stop cardiorespirator, paralizie a corzilor vocale și slăbiciune a mâinii în decurs de 2 săptămâni de la debutul clinic a fost de 71%. Cu o deteriorare mai treptată, mortalitatea a fost de 23%. Cel mai prost factor de prognostic în raport cu eficacitatea tratamentului chirurgical a fost paralizia bilaterală a corzilor vocale.
Alte tipuri de malformații Chiari
Malformația Chiari tip 3
Apare rar. Este cea mai severă formă. Deplasarea structurilor PCF cu hernie a cerebelului prin BZO în canalul cervical, adesea cu encefalomeningocel cervical sau suboccipital. De obicei incompatibil cu viața.
Malformația Chiari tip 4
Hipoplazia cerebelului fără hernie a cerebelului.
Greenberg. Neurochirurgie
La sfârșitul secolului al XIX-lea, anomalia a fost descrisă pentru prima dată de patologul austriac Hans Chiari, care în secolul următor a fost numit „Sindromul Arnold-Chiari”. Malformația este o patologie congenitală.
Ce este sindromul Arnold-Chiari?
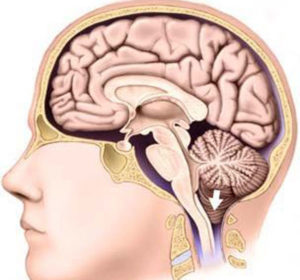
Cu o astfel de anomalie, întreaga structură a fosei posterioare a craniului cade sub foramen magnum.
BZO este zona de limită dintre măduva spinării și creier. În absența patologiei, lichidul cefalorahidian sau lichidul cefalorahidian se mișcă liber în spațiul subarahnoidian. Cu sindromul Arnold-Chiari amigdalele cerebeloase înfundă deschiderea, perturbând astfel fluxul de lichid cefalorahidian, ceea ce contribuie la formarea hidrocefaliei.
Tipuri și grade ale bolii
Sindromul Arnold-Chiari are trei tipuri − I, II și III, IV. Toate sunt asociate cu o localizare subestimată a amigdalelor cerebeloase.
La patologie generală, tipurile de MK au anumite diferențe:
- Sindromul de tip I reprezinta omiterea amigdalelor si deplasarea creierului mic sub BZO. Cu acest tip de anomalie, fosa craniană posterioară este mică.
La diagnosticare, pot fi distinse trei variante ale sindromului - anterioară, intermediară și posterioară:
- Cu optiunea fata există o deplasare a C2 înapoi, atârnând peste procesul odontoid, amprenta bazilară și platybasia;
- Dacă există o opțiune intermediară există compresie de către amigdalele deplasate ale cerebelului medulei alungite ventrale și segmentelor superioare ale măduvei spinării;
- Varianta spate duce la comprimarea secțiunilor cervicale superioare ale secțiunilor spinale și dorsale ale medulei oblongate.
Primul tip de malformație Chiari considerată de neurologi cea mai frecventă boală. Mai des decât altele, suferă de femei care au născut târziu și au intrat în perioada premenopauză.
- Sindromul de tip II are o formă congenitală. Progresia patologiei are loc în timpul dezvoltării fetale. O anomalie poate fi detectată în timpul examinării fătului. Medula oblongata, ventriculul IV, partea inferioară a viermelui sunt înțepate la 2 mm sub intrarea în foramenul mare posterior.
- Se caracterizează tipul III de patologie coborâre a micului și medular oblongata până la meningocelul regiunii cervico-occipitale.
- IV tip de patologie este o subdezvoltare a cerebelului fără coborâre a amigdalelor în BZO.
La începutul secolului XXI, a tip nul de anomalie. Este determinată de prezența hidromieliei fără amigdalele scăzute ale creierului mic.
Atenţie! Malformația Arnold-Chiari de tip IV a început să fie atribuită patologiei Dandy-Walker.
Elena Potemkina, după o serie de studii, a propus să clasifice sindromul în funcție de gradul de coborâre a amigdalelor cerebeloase în foramen magnum:
- 0 grad- amigdalele sunt la nivelul superior al 3D-ului;
- 1 grad- amigdalele s-au deplasat spre marginea superioară a arcului atlasului sau C1;
- 2 grade– la nivelul marginii superioare a C2;
- 3 grade- la nivelul varfului C3 si mai jos, in sens caudal.
Al doilea și al treilea fel sindromul se manifestă împreună cu alte boli ale centralei sistem nervos. Al treilea și al patrulea incompatibil cu viata.
Simptome
Semnele anomaliei Arnold-Chiari pot fi atât independente, cât și generale.
Acestea includ următoarele simptome merită să acordați atenție:
- La întoarcerea capului și în repaus un sunet de creștere neobișnuit se aude în urechi sub formă de zgomot, zumzet, fluier;
- La activitate fizica din cauza tensiunii durere de cap;
- TIC nervos globii oculari sau nistagmus;
- Dureri la nivelul occiputului dimineața. A ridica interior presiunea craniană, amorțeală;
- Din cauza disfuncției cerebelului pot apărea amețeli și dezorientare;
- În mișcare, există neîndemânare;
- Cu o formă severă a bolii există un tremur al membrelor;
- Apar probleme de vedere sub forma unui „voal în fața ochilor”, bifurcare a obiectelor, orbire. La întoarcerea capului, fenomenele se intensifică. Acest lucru se datorează presiunii asupra medulului oblongata;
- Mușchii membrelor, fețele și trunchiul devin mult mai slabe;
- Probleme cu urinarea observat în formă avansată și severă a bolii;
- Pragul de sensibilitate devine mai puțin într-una dintre părțile corpului sau a feței, membre;
- La viraje ascuțite Capete pacientul își poate pierde cunoștința;
- Perturbare la locul de muncă sistemul respirator - respirație grea sau oprirea ei completă;
- Complicații severe- paralizia membrelor, hidrocefalie, formarea de chisturi la nivelul coloanei vertebrale, deformarea oaselor piciorului.
semn distinctiv sindromul de tip I este o durere de cap persistentă. La tip II, care se manifestă imediat după naștere sau în copilăria timpurie, se pot observa probleme la înghițire, un plâns slab.
Toate simptomele de mai sus se agravează odată cu mișcarea.
Atenţie! Cu acces prematur la un medic și lipsa tratamentului, poate apărea un atac de cord al creierului sau măduvei spinării.
Cauzele anomaliei
Oamenii de știință nu au înțeles încă pe deplin cauzele sindromul Arnold-Chiari. Malformația Chiari, așa cum este numită și anomalia, se poate dezvolta din cauza mai multor factori.
Congenital:
- Dezvoltarea anormală a oaselor craniului la făt- fosa posterioară prea mică a craniului la un bebeluș care se dezvoltă corespunzător duce la o discrepanță între creșterea oaselor craniului și a creierului. Doar că oasele craniene nu pot ține pasul cu creierul.
- Fătul este BZO hipercrescut.
Cumparat:
- În timpul nașterii a avut loc o leziune cerebrală traumatică;
- din cauza tulburărilor circulatorii livkor în ventriculii creierului provoacă leziuni ale măduvei spinării. Se ingroasa.
Sindromul Arnold-Kiara poate apărea simultan sau după alte defecte, de exemplu - hidropizie a măduvei spinării.
Nu cu mult timp în urmă, experții credeau că această boală este congenitală, dar procentul de pacienți cu o anomalie congenitală este mult mai mic decât procentul cu dobândit sindrom.
Factori de risc
Riscul de a dezvolta o malformație la făt poate fi crescut de factori precum: 
- necontrolat utilizarea medicamentelor în timpul sarcinii;
- Disponibilitate obiceiuri proaste la o femeie însărcinată;
- Transferat mama bebelușului boli infecțioase. Cum ar fi rubeola și altele;
- Problemă din alimentatie buna la o femeie însărcinată.
Majoritatea experților sunt de părere că sindromul Arnold-Chiari apare pe fondul mutațiilor genetice la un copil. Legături cu anomalie cromozomiala pentru moment nu a fost gasit.
Sindromul Arnold-Chiari la făt
Cu o examinare cu ultrasunete, medicul poate detecta prezența patologiei în partea din spate a creierului. După primul trimestru de sarcină cu ecografie trebuie să fie vizibile contururile emisferelor cerebrale ale bebelușului cu creierul mic în curs de dezvoltare. Dimensiunile și formele neclare ale creierului pot indica un sindrom.
Ultrasunetele pot fi folosite pentru a detecta malformații de primul și al doilea tip. Pe lângă această metodă de cercetare, se utilizează ecografie. Poate fi folosit pentru a detecta ventriculii ascuțiți laterali. corect diagnostic instrumental fătul ajută la stabilirea unui diagnostic precis.
Examinare și diagnosticare
Se întâmplă că Patologia Chiari nu se manifestă, ci se găsește numai la examinarea unui medic. O examinare generală folosind EEG, REG și ECHO-EG poate evidenția creșterea presiunii intracraniene. O radiografie va arăta dacă există sau nu anomalii în oasele craniului. Dar diagnosticul final cu o astfel de verificare este imposibil de făcut.
Dacă sunteți sigur că există o patologie, specialistul va trimite pacientul la imagistică prin rezonanță magnetică măduva spinării și creierul. În timpul studiului, pacientului i se interzice deplasarea. Sugarii și copiii mici sunt cufundați în el somn medical ceea ce va ajuta la realizarea cu succes a anchetei. Poate fi necesar un agent de contrast pentru a obține o imagine clară.
Pentru a exclude prezența siringomieliei, prescrieți tomografieîntreaga coloană vertebrală.
Complicații
Fără tratament lipsit sau inadecvat sindromul Arnold-Chiari pot apărea paralizii ale nervilor cranieni. Formați un chist pe coloana vertebrală. Patologia contribuie la apariția hidrocefaliei. Din această cauză, lichidul se va acumula în creier. Pentru drenarea lichidului cefalorahidian, medicii vor recurge la manevră și drenare.
Diagnosticul și tratamentul târziu pentru sindromul Arnold-Chiari vor duce la consecințe grave cu un rezultat letal.
Tratament
Dacă se detectează o anomalie, tratamentul trebuie început imediat. Boala primele două tipuri predispus la terapie.
metoda conservatoare
Pentru a prescrie un curs de tratament, medicul trebuie să studieze toți indicatorii examinării pacientului. Dacă simptomul principal al bolii este o durere de cap, atunci va fi prescris un tratament conservator.
Medicamente antiinflamatoare nesteroidiene, relaxante musculare vor fi prescrise:
După eliminare sindrom de durere numit diuretice. Puteți elimina răgușeala cu ajutorul cursurilor cu un logoped. cu exceptia tratament medicamentos exercițiu util.
Cursul terapiei durează câteva luni, după care este necesar să se supună examen complet. Cu absenta rezultate pozitive tratament chirurgical prescris.
Intervenție chirurgicală
Dacă terapie conservatoare nu a adus rezultate pozitive sau pacientul are simptome grave, apoi i se prescrie o laminectomie, craniectomie decompresivă a fosei posterioare. La fel și plasticitatea învelișului dur al creierului. Această operație vă permite să creșteți volumul fosei craniene și să extindeți foramenul magnum. Toate acestea vor duce la încetarea stoarcerii, îmbunătățind fluxul de lichid cefalorahidian.
Daca este disponibil hidrocefalie, apoi ventriculul este retras folosind o supapă specială.
În timpul operațiunii, utilizați control electrofiologic, cu care poți decide să deschizi sau nu întreaga dura mater.
curs de reabilitare după intervenție chirurgicală ia sapte zile.
Important! Pacienții cu sindrom Arnold-Chiari, în ciuda tratamentului, ar trebui să viziteze periodic un medic.
malformatie Chiari nu este pe deplin înțeles și nu există nicio modalitate de a preveni complet dezvoltarea acestei boli. Dar viitorii părinți, și mai ales mamele, pot reduce la maximum riscul de patologie. Pentru aceasta, este necesar să imagine corectă viața, studiați toți factorii de risc de mai sus și eliminați-i dacă este posibil.
Sănătatea copilului tău depinde de tine.
Anomalia Arnold-Chiari este o încălcare a structurii și locației cerebelului, a trunchiului cerebral în raport cu craniul și canalul spinal. Această condiție se referă la defecte congenitale dezvoltare, deși nu se manifestă întotdeauna încă din primele zile de viață. Uneori, primele simptome apar după 40 de ani. Anomalia Arnold-Chiari se poate manifesta prin diferite simptome de afectare a creierului, măduvei spinării, circulație afectată a lichidului cefalorahidian. Încheierea diagnosticului este de obicei pusă prin imagistica prin rezonanță magnetică. Tratamentul este conservator și metode chirurgicale. Din acest articol puteți afla mai multe despre simptomele, diagnosticul și tratamentul anomaliei Arnold-Chiari.
În mod normal, limita dintre cap și este la nivelul dintre oasele craniului și coloana cervicală. Aici este un foramen occipital mare, care, de fapt, servește ca o linie condiționată. Condițional, deoarece țesutul cerebral trece în măduva spinării fără întrerupere, fără o limită clară. Toate structurile anatomice situate deasupra foramenului magnum, în special, medulara oblongata, puntea și cerebelul, aparțin formațiunilor fosei craniene posterioare. Dacă aceste formațiuni (una câte una sau toate împreună) coboară sub planul foramenului magnum, atunci apare anomalia Arnold-Chiari. O astfel de locație incorectă a cerebelului, medulla oblongata duce la compresia măduvei spinării în zonă cervicale coloanei vertebrale, interferează cu circulația normală a lichidului cefalorahidian. Uneori, anomalia Arnold-Chiari este combinată cu alte malformații ale joncțiunii craniovertebrale, adică locul unde craniul trece în coloana vertebrală. În astfel de cazuri combinate, simptomele sunt de obicei mai pronunțate și se fac simțite destul de devreme.
Anomalia Arnold-Chiari poartă numele a doi oameni de știință: patologul austriac Hans Chiari și patologul german Julius Arnold. Prima, în 1891, a descris o serie de anomalii în dezvoltarea cerebelului și a trunchiului cerebral, a doua, în 1894, a oferit o descriere anatomică a coborârii părții inferioare a emisferelor cerebeloase în foramen magnum. .
Varietăți ale anomaliei Arnold-Chiari
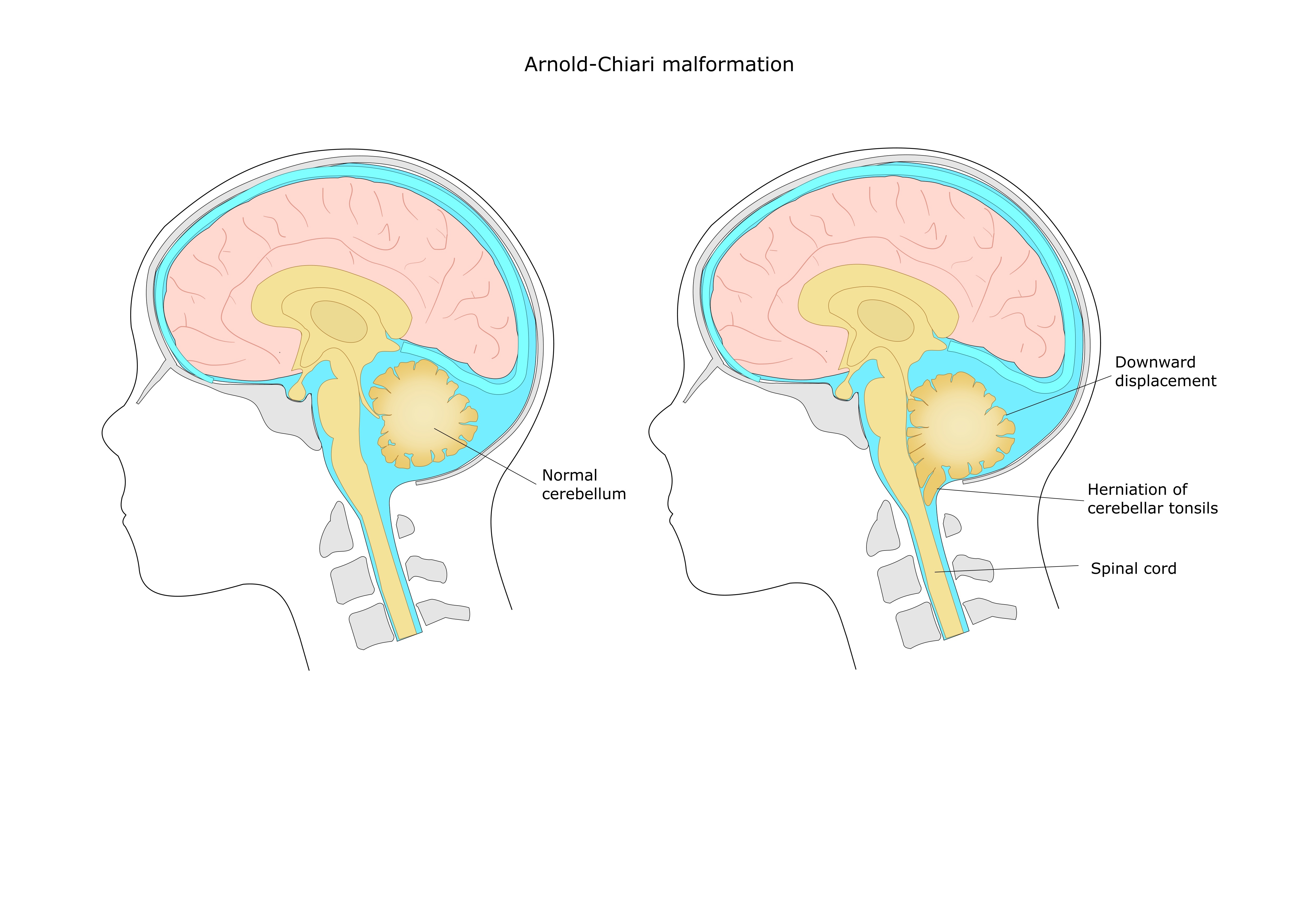
Conform statisticilor, anomalia Arnold-Chiari apare cu o frecvență de 3,2 până la 8,4 cazuri la 100.000 de locuitori. O gamă atât de largă se datorează parțial eterogenității acestei malformații. Despre ce este vorba? Faptul este că anomalia Arnold-Chiari este de obicei împărțită în patru subtipuri (descrise de Chiari), în funcție de ce structuri sunt coborâte în foramen magnum și cât de neregulate sunt în structură:
- anomalie Arnold-Chiari I - când amigdalele cerebeloase coboară în canalul spinal din craniu (partea inferioară a emisferelor cerebeloase);
- Anomalie Arnold-Chiari II - când coboară în canalul rahidian majoritatea cerebel (inclusiv viermele), medular oblongata, ventricul IV;
- Anomalie Arnold-Chiari III - când aproape toate formațiunile fosei craniene posterioare (cerebel, medular oblongata, ventricul IV, punte) sunt situate sub foramen magnum. Destul de des sunt localizate în hernia cerebrală a regiunii cervico-occipitale (o situație în care există un defect al canalului spinal sub forma unei despicaturi a arcurilor vertebrale și conținutul sacului dural, adică măduva spinării cu toate membranele, iese în acest defect). Diametrul foramenului magnum în cazul acestui tip de anomalie este crescut;
- Anomalie Arnold-Chiari IV - subdezvoltarea (hipoplazia) a cerebelului, dar cerebelul în sine (sau mai bine zis, ceea ce s-a format în locul său) este localizat corect.
Tipurile I și II de defecte sunt mai frecvente. Acest lucru se datorează faptului că tipurile III și IV nu sunt de obicei compatibile cu viața, moartea survine în primele zile de viață.
Până la 80% din toate cazurile de anomalie Arnold-Chiari sunt combinate cu prezența siringomieliei (o boală caracterizată prin prezența unor cavități în măduva spinării care înlocuiesc țesutul cerebral).
În dezvoltarea anomaliei, rolul principal revine tulburărilor în formarea structurilor creierului și coloanei vertebrale în perioada prenatală. Cu toate acestea, trebuie luat în considerare și următorul factor: o leziune la cap suferită în timpul nașterii, leziuni cranio-cerebrale repetate în copilărie poate deteriora suturile osoase de la baza craniului. Ca urmare, formarea normală a fosei craniene posterioare este perturbată. Devine prea mic, cu o pantă aplatizată, din cauza căreia toate structurile fosei craniene posterioare pur și simplu nu sunt capabile să se potrivească în ea. Ei „căută o cale de ieșire” și se grăbesc în foramenul occipital mare și apoi în canalul spinal. Această situație este într-o oarecare măsură considerată o anomalie Arnold-Chiari dobândită. De asemenea, simptome similare cu anomalia Arnold-Chiari pot apărea odată cu dezvoltarea unei tumori pe creier, care face ca emisferele cerebeloase să se deplaseze în foramen magnum și canalul spinal.
Simptome
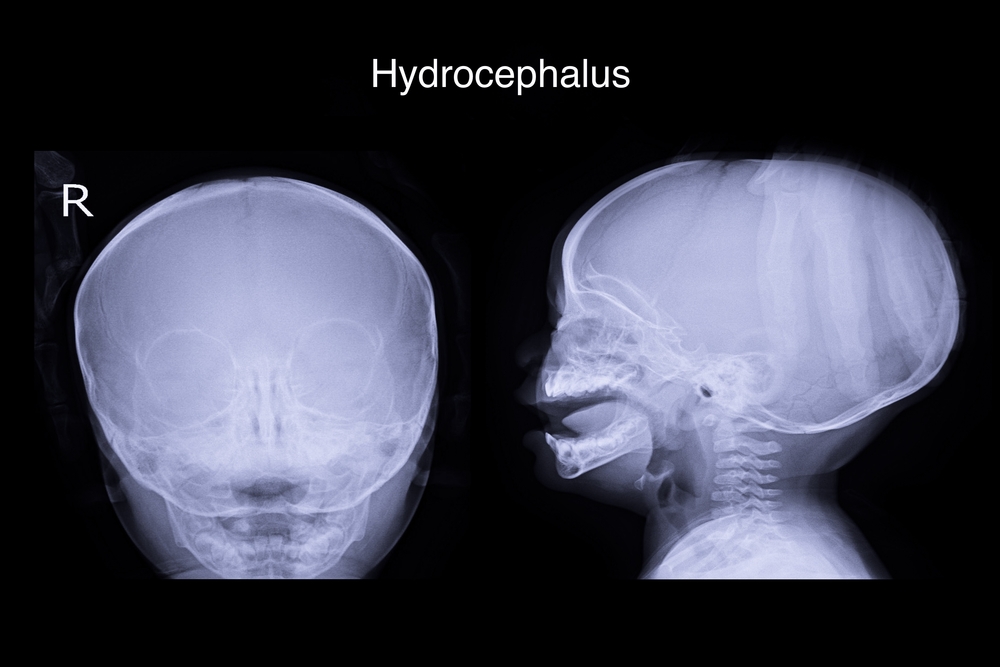 Una dintre manifestările anomaliei Arnold-Chiari poate fi hidrocefalia.
Una dintre manifestările anomaliei Arnold-Chiari poate fi hidrocefalia. Principalele manifestări clinice ale anomaliei Arnold-Chiari sunt asociate cu compresia structurilor creierului. În același timp, vasele care hrănesc creierul, căile de curgere a lichidului și rădăcinile nervilor cranieni care trec în această zonă sunt comprimate.
Se obișnuiește să se distingă 6 sindroame neurologice care pot însoți anomalia Arnold-Chiari:
- cerebelos;
- bulbar-piramidal;
- radicular;
- siringomielitic.
Desigur, nu sunt întotdeauna prezente toate cele 6 sindroame. Severitatea lor variază într-un grad sau altul, în funcție de ce structuri sunt comprimate și cât de mult.
Hipertensiv-hidrocefalic sindromul se dezvoltă ca urmare a unei încălcări a circulației lichidului cefalorahidian (LCR). În mod normal, LCR curge liber din spațiul subarahnoidian al creierului către spațiul subarahnoidian al măduvei spinării. Partea inferioară descendentă a amigdalelor cerebelului blochează acest proces, ca un dop de plută într-o sticlă. Formarea lichidului cefalorahidian în plexurile vasculare ale creierului continuă și, în general, nu există unde să curgă (fără a lua în calcul mecanismele naturale de absorbție, care nu sunt suficiente în acest caz). LCR se acumulează în creier, determinând creșterea presiunii intracraniene (hipertensiune intracraniană) și extinderea spațiilor care conțin LCR (hidrocefalie). Aceasta se manifestă ca o durere de cap explozivă, care este agravată de tuse, strănut, râs, încordare. Durerea se simte în partea din spate a capului, în zona gâtului, este posibilă tensiunea musculară a gâtului. Pot exista episoade de vărsături bruște, în niciun caz legate de aportul alimentar.
cerebeloasă sindromul se manifestă ca o încălcare a coordonării mișcărilor, un mers „beat” și o ratare atunci când se efectuează mișcări intenționate. Pacienții sunt îngrijorați de amețeli. Poate exista tremur la nivelul membrelor. Vorbirea poate fi perturbată (devine împărțită în silabe separate, cântând). Un simptom destul de specific este considerat a fi „nistagmusul care lovește”. Acestea sunt zvâcniri involuntare ale globilor oculari, îndreptate, în acest caz, în jos. Pacienții se pot plânge de vedere dublă din cauza nistagmusului.
Bulbar-piramidal sindromul este numit astfel pentru denumirea structurilor care sunt supuse compresiunii. Bulbus este numele medulei oblongata din cauza formei sale bulboase, astfel încât sindromul bulbar înseamnă semne de deteriorare a medulului oblongata. Iar piramidele sunt formațiunile anatomice ale medulei oblongate, care sunt mănunchiuri de fibre nervoase care transportă impulsuri din cortex. emisfere la celule nervoase coarnele anterioare ale măduvei spinării. Piramidele sunt responsabile pentru mișcările voluntare ale membrelor și trunchiului. În consecință, sindromul bulbar-piramidal se manifestă clinic ca slăbiciune musculară la nivelul membrelor, amorțeală și pierderea durerii și a sensibilității la temperatură (fibrele trec prin medula oblongata). Comprimarea nucleilor nervilor cranieni localizați în trunchiul cerebral provoacă tulburări de vedere și auz, vorbire (datorită deficienței mișcărilor limbii), voce nazală, sufocare când mănâncă și dificultăți de respirație. Poate exista pierderea pe termen scurt a conștienței sau pierderea tonusului muscular cu starea de conștiență păstrată.
Rădăcină sindromul in cazul anomaliei Arnold-Chiari consta in aparitia semnelor de disfunctie a nervilor cranieni. Acestea pot fi mobilitate redusă a limbii, voce nazală sau răgușită, tulburări de deglutiție, defecte de auz (inclusiv tinitus), tulburări senzoriale ale feței.
Sindromul de insuficiență vertebrobazilară asociat cu aportul de sânge afectat în bazinul de sânge corespunzător. Din această cauză apar atacuri de amețeli, pierderea conștienței sau tonusului muscular și probleme de vedere. După cum puteți vedea, devine clar că majoritatea simptomelor anomaliei Arnold-Chiari apar nu ca urmare a unei cauze directe, ci datorită influenței combinate. diverși factori. Astfel, atacurile de pierdere a cunoștinței sunt cauzate atât de comprimarea unor centri specifici ai medulei oblongate, cât și de afectarea aportului de sânge în bazinul vertebrobazilar. O situație similară apare cu tulburări de vedere, auz, amețeli și așa mai departe.
Siringomielitic sindromul nu apare întotdeauna, ci numai în cazurile unei combinații de anomalie Arnold-Chiari cu modificări chistice la nivelul măduvei spinării. Aceste situații se manifestă printr-o încălcare disociată a sensibilității (atunci când temperatura, durerea și sensibilitatea tactilă sunt perturbate izolat, iar adâncimea (poziția membrului în spațiu) rămâne intactă), amorțeală și slăbiciune musculară la unele membre, disfuncția pelvină. organe (incontinență urinară și fecală). Despre cum se manifestă siringomielia, puteți citi într-un articol separat.
Fiecare tip de anomalie Arnold-Chiari are propriile sale caracteristici clinice. Anomalia de tip I Arnold-Chiari poate să nu se manifeste în niciun fel până la vârsta de 30-40 de ani (în timp ce corpul este tânăr, comprimarea structurilor este compensată). Uneori, acest tip de defect este o descoperire accidentală în timpul imagistică prin rezonanță magnetică pentru o altă boală.
Tipul II este adesea combinat cu alte defecte: meningomielocel regiunea lombară si stenoza apeductului cerebral. Manifestările clinice apar încă din primele minute de viață. În plus față de simptomele principale, copilul are respirație puternică cu perioade de oprire, o încălcare a înghițirii laptelui, alimente care intră în nas (copilul se sufocă, se sufocă și nu poate suge sânul).
Tipul III este adesea combinat cu alte malformații ale creierului și ale regiunii cervico-occipitale. Într-o hernie cerebrală în regiunea cervical-occipitală, nu numai cerebelul, ci și medulara oblongata, pot fi localizați lobii occipitali. Acest defect este practic incompatibil cu viața.
Tipul IV de către unii oameni de știință, recent, nu este considerat un complex de simptome Chiari în vedere modernă, deoarece nu este însoțită de coborârea cerebelului subdezvoltat în foramen magnum. Totuși, clasificarea austriacului Chiari, care a descris pentru prima dată această patologie, conține și tipul IV.
Diagnosticare
Combinația unui număr de simptome descrise mai sus permite medicului să suspecteze o anomalie Arnold-Chiari. Dar pentru confirmarea exactă a diagnosticului, este necesar să se efectueze imagistica prin rezonanță magnetică sau computerizată (aceasta din urmă metodă este mai informativă). Imaginea RMN arată coborârea structurilor fosei craniene posterioare sub foramen magnum și confirmă diagnosticul.
Tratament
Alegerea tratamentului pentru anomalia Arnold-Chiari depinde de prezența simptomelor bolii.
Dacă defectul a fost detectat întâmplător (adică nu manifestari cliniceși nu deranjează pacientul) atunci când se efectuează imagistica prin rezonanță magnetică pentru o altă boală, atunci tratamentul nu se efectuează deloc. Pacientul este monitorizat dinamic pentru a nu rata momentul aparitiei primului simptome clinice compresia creierului.
Dacă anomalia se manifestă ca un sindrom hipertensiv-hidrocefalic ușor pronunțat, atunci se încearcă tratament conservator. În acest scop utilizați:
- medicamente pentru deshidratare (diuretice). Acestea reduc cantitatea de lichid cefalorahidian, ajutând la reducerea durerii;
- medicamente antiinflamatoare nesteroidiene pentru a reduce durerea;
- relaxante musculare în prezența tensiunii musculare în regiunea cervicală.
Dacă aplicațiile medicamente se dovedește a fi suficient, apoi pentru o perioadă se opresc acolo. Dacă nu există niciun efect sau pacientul are semne ale altor sindroame neurologice ( slabiciune musculara, pierderea senzației, semne de disfuncție a nervilor cranieni, atacuri periodice de pierdere a cunoștinței și așa mai departe), apoi recurg la tratament chirurgical.
Tratamentul chirurgical constă în trepanarea fosei craniene posterioare, îndepărtarea unei părți a osului occipital, rezecția amigdalelor cerebeloase coborâte în foramen magnum, disecția aderențelor spațiului subarahnoidian care interferează cu circulația LCR. Uneori poate fi necesară o intervenție chirurgicală de bypass pentru a drena excesul de lichid cefalorahidian. „Lichidul în exces” este evacuat printr-un tub special (șunt) în piept sau cavitate abdominală. Stabilirea când apare nevoia tratament chirurgical, este o sarcină foarte importantă și responsabilă. Modificările pe termen lung ale sensibilității, pierderea forței musculare și defectele nervilor cranieni pot să nu se recupereze complet după intervenție chirurgicală. Prin urmare, este important să nu ratați momentul în care cu adevărat nu vă puteți descurca fără intervenție chirurgicală. Cu defect de tip II tratament chirurgical prezentate în aproape 100% din cazuri fără tratament conservator prealabil.
Astfel, anomalia Arnold-Chiari este una dintre malformațiile dezvoltării umane. Poate fi asimptomatică sau se poate manifesta încă din primele zile de viață. Manifestările clinice ale bolii sunt foarte diverse, diagnosticul se realizează folosind imagistica prin rezonanță magnetică. Abordările de tratament variază de la fără intervenție la metode operaționale. Volum măsuri medicale determinată individual.
Fotografie de la Barcelona Chiari & Syringomyelia & Scoliosis Institute institutchiaribcn.com/en/
Boala Arnold-Chiari aparține unei patologii ereditare, totuși, semnele inițiale poate apărea la vârsta adultă (după 40 de ani). Baza bolii este discrepanța dintre poziția medulei oblongate, cerebelul față de foramenul cranian mare și încălcarea structurii acestora. Această patologie se caracterizează prin prezența unui număr de tulburări neurologice. Luați în considerare de ce apare boala, cum se manifestă clinic și cum să tratați pacienții.
Posibile tipuri de boală și factori provocatori
În mod normal, deschiderea mare a craniului servește ca o limită condiționată între structurile craniului (cerebel, medula oblongata, pons), măduva spinării. Boala Arnold-Chiari este diagnosticată la coborârea unor părți ale creierului sub această limită, compresia (compresia) acestora și apariția simptomelor corespunzătoare. Există mai multe subtipuri de boală:
- Patologia de tip 1 Arnold-Chiari - din cauza deplasării medulei oblongate, amigdalele cerebeloase, care alcătuiesc partea inferioară a emisferelor sale, sunt localizate în canalul rahidian sub foramenul cranian. Boala este detectată la vârsta adultă sau poate fi o constatare incidentală în timpul studiului din cauza unui alt diagnostic.
- Patologia Arnold-Chiari de tip 2 - mai multe structuri ale creierului (o parte semnificativă a cerebelului, ventriculului IV, medular oblongata) se află în canalul spinal. Această subspecie a bolii este adesea combinată cu manifestări în îngustarea apeductului cerebral, hernie lombar măduva spinării și siringomielie stare patologică când se formează carii – chisturi – în locul țesutului cerebral. Cel mai adesea se găsește la bebeluși în prezența manifestărilor clinice.
- Patologia tipurilor 3 și 4 nu oferă practic nicio șansă de supraviețuire, iar un bebeluș bolnav moare în primele zile de viață.
De asemenea, clasificată ca anomalie Arnold-Chiari de gradul I - descrisă ca boala ereditara si se transmite copiilor de la parinti bolnavi (rude). O boală rezultată din cauza unei leziuni intrauterine infecțioase a creierului este denumită anomalie Arnold-Chiari de gradul 2.
Există mai multe teorii cu privire la motivul pentru care apare boala. Să aducem motive posibile boli:
- factor ereditar.
- Defecte congenitale ale craniului - se formează partea sa occipitală prea mică, care fizic nu poate găzdui toate structurile necesare ale creierului. Ei, crescând în dimensiune, pun presiune pe deschiderea craniului, trec în canalul spinal.
- Cursul patologic al sarcinii și afectarea creierului copilului sub influența infecțiilor materne, adesea recurente boli virale, doze mari droguri, alcool, compuși toxici și nicotină.
- Leziuni primite de copil în timpul nașterii și re-rănirea capului în vârstă fragedă poate rupe suturile craniene.
Important! Neoplasmele creierului de dimensiuni considerabile duc la o creștere a presiunii în interiorul craniului și indirect „forțează” o parte a cerebelului să se deplaseze sub deschiderea craniului. Astfel, există o boală similară cu patologia descrisă.
Manifestări de bază și sindroame neurologice
 Simptomele anomaliei Arnold-Chiari sunt grupate în mai multe sindroame neurologice, în funcție de severitatea cursului și de afectarea maximă a anumitor componente ale creierului:
Simptomele anomaliei Arnold-Chiari sunt grupate în mai multe sindroame neurologice, în funcție de severitatea cursului și de afectarea maximă a anumitor componente ale creierului:
- O creștere a presiunii în interiorul craniului se formează atunci când circulația liberă și fluxul de LCR sunt perturbate. Există o amenințare de hidrocefalie, care este detectată clinic prin dureri de cap, acestea sunt resimțite în ceafă, ceafă, agravată atunci când pacientul râde sau tusește. Adesea există vărsături, care nu sunt asociate cu mesele.
- Sindromul cerebelos se manifestă prin tulburări ale mersului și coordonării în spațiu, pacientul nu poate efectua mișcări precise, mâinile îi tremură, iar capul se învârte. Pacientul vorbește în silabe separate, cântând fiecare cuvânt. Există, de asemenea, probleme de vedere, vedere dublă, pupile decăzute (nistagmus).
- Afectarea medulei oblongate este descrisă de prezența sindromului bulbar-piramidal. Pacientul are slăbiciune musculară, membrele amorțesc, durerea și sensibilitatea la temperatură se pierde. Vocea devine nazală, există dificultăți de respirație și de a lua apă și mâncare. Deteriorarea nucleilor nervilor craniului amenință cu afectarea auzului, a vorbirii și a vederii.
- Tulburările de funcționare a limbii, deglutiția, modificările vocii și sensibilitatea pielii feței sunt combinate într-un sindrom radicular.
- Deteriorarea alimentării cu sânge a creierului se caracterizează prin amețeli, o scădere a tonusului muscular și pierderea conștienței, tulburări de vedere și sunt incluse în sindrom.
Managementul pacientului
Tratamentul anomaliei Arnold-Chiari depinde de severitatea cursului și de plângerile pacientului. Sunt posibile următoarele tactici terapeutice:
- în absența manifestărilor și plângerilor clinice - observarea pacientului;
- tratament medicamentos pentru reducerea presiunii craniene, analgezice, medicamente pentru normalizarea tonusului muscular;
- intervenție chirurgicală pentru restabilirea fluxului liber al lichidului cefalorahidian și reducerea presiunii prin șuntare

